
Interpretable Meta-Learning for Multi-Objective Chemical Search
Source: arXiv:2606.20497 · Published 2026-06-18 · By Antonio Varagnolo, Yulia Pimonova, Michael G. Taylor, Raphaël Pestourie, Nicholas E. Lubbers
TL;DR
This work tackles the challenge of multi-objective molecular discovery under computational restrictions and data scarcity by introducing an interpretable and data-efficient meta-learning framework integrated into an active search pipeline. The authors propose using linear graphlet-based meta-learning surrogates trained across multiple chemical property prediction tasks, combined with adaptive uncertainty quantification via Bayesian bootstrap ensembles and an Efficient Global Optimization loop. This approach enables rapid transfer learning to new objectives from limited data while preserving interpretability and computational efficiency.
Empirical evaluation on the QM9 benchmark and a challenging real-world spin-crossover (SCO) metal-organic complex search shows that meta-learning surrogates substantially outperform baseline linear models and random search. The meta-learned surrogates dominate baseline Pareto fronts by 78% in SCO multi-objective optimization and accelerate convergence by about two orders of magnitude on QM9. The authors also demonstrate robust calibration improvements through a novel adaptive confidence tuning strategy, which further improves Pareto front quality over static uncertainty calibration. An ablation confirms that meta-learning acts as a chemically aware structural bias, increasing feature utilization and reducing overfitting under scarce data. This constitutes the first application of linear meta-learning in multi-objective chemical search paired with dynamic uncertainty calibration.
Key findings
- Meta-learning surrogate models dominate baseline linear regression Pareto fronts in spin-crossover complex search by 78% on average (Fig. 5a).
- Meta-learning accelerates convergence in QM9 benchmark by roughly two orders of magnitude compared to random sampling (Fig. 4).
- RMSE reduction of up to 47% for meta-learning surrogates vs. base linear models on SCO candidates with only 2× computational cost.
- Dynamic confidence tuning improves Pareto front dominance over static calibration in SCO search, dominating 52% of the static front (Fig. 5b).
- Meta-learning increases number of molecular subgraphs contributing to predictions by approximately an order of magnitude under low-data conditions.
- Bayesian bootstrap ensembles enable calibrated, non-Gaussian uncertainty estimates robust to distribution shift during iterative search.
- Pipeline evaluates ~60000 proposed molecules per iteration, clusters top 2000 candidates, and selects 399 representatives, balancing exploration and exploitation.
- Meta-learning surrogates outperform base models on 91% of evaluated SCO candidates during search.
Methodology — deep read
Threat Model & Assumptions: The adversary is implicitly the intractability and expense of exploring vast molecular spaces with conflicting objectives and scarce labeled data. No explicit attacker modeled. Assumptions include limited labeled data for new objectives, distribution shift due to tail candidates, and computational constraints requiring interpretable, fast surrogates.
Data: Used QM9 dataset (134k small organic molecules with DFT properties) for offline benchmarking across atomization energy, zero-point energy, electronic gap, and heat capacity. Also performed a large-scale online search for octahedral iron spin-crossover complexes using a filtered ligand set from the Cambridge Structural Database. Ligands are functionalized systematically to generate candidate complexes, which are constructed and evaluated for spin-state energies and solvation properties using Architector and XTB quantum chemistry tools. Some candidate evaluations fail due to geometry build errors, reducing effective dataset size.
Architecture/Algorithm: Core surrogate models are linear ridge regressions over sparse high-dimensional (∼10k feature) graphlet fingerprint vectors representing molecular subgraphs. The authors apply the LAMeL linear meta-learning framework, which learns a low-dimensional subspace encoding shared chemical knowledge across multiple support tasks (both search objectives and auxiliary cheap-to-compute properties such as solubility). Specialization tasks (new objectives) are modeled as adapted linear combinations of support coefficients plus residual corrections, enabling rapid adaptation from limited data. Uncertainty is quantified by Bayesian bootstrapping, generating an ensemble by fitting models with Dirichlet-weighted training samples, producing calibrated predictive distributions capturing distributional mismatch and empirical uncertainty. Candidate acquisition leverages an Efficient Global Optimization framework balancing exploration-exploitation via Probability of Improvement (PoI) applied to ensemble surrogates. Sampling draws from Pareto-optimal molecules with novel functionalizations and diverse ligand pools. Clustering via K-means reduces candidate sets for evaluation. Adaptive confidence tuning dynamically adjusts the Dirichlet concentration parameter α controlling bootstrap variance to recalibrate uncertainty estimates using feedback from log-normalized squared error statistics. This prevents overconfident or underconfident predictions during the evolving, non-stationary search distribution.
Training Regime: Each iteration fits multiple ridge regression models in parallel (e.g., B=100 bootstrap resamples times No=number of objectives plus Na auxiliary tasks) with regularization. Meta-learning subspace is constructed from support task coefficients. Exact hyperparameters like ridge alpha not specified. Number of iterations ranges from few (QM9) to ~50 for SCO search.
Evaluation Protocol: Empirical evaluation via multiple independent runs (five seeds) comparing random, baseline linear, and meta-learning guided search strategies. Metrics include dominance-based c-metric comparing Pareto fronts (fraction of points dominated), marginal improvements in single objectives, and RMSE on predictions. Ablations examine feature utilization and sparsity. Calibration is assessed via log-normalized squared error φ(t). Scalability demonstrated by cluster-selected candidate evaluation batches.
Reproducibility: Code availability and dataset openness not explicitly stated. QM9 is public, SCO ligand sets come from CSD with chemical filters. Specific parameter choices and computational infrastructure details partially described but not fully detailed. Overall methodology is modular and parallelizable.
Example Workflow End-to-End: Starting with an initial batch of M=500 evaluated SCO ligands, the pipeline fits B×No+Na ridge regressions forming meta-learned surrogate models with uncertainty via Bayesian bootstrap ensembles. P=60000 candidate complexes are generated by functionalizing Pareto-optimal ligands and sampling filtered ligand pools. These candidates are ranked with PoI acquisition scores, the top S=2000 are clustered into K=399 representatives, which are then evaluated with Architector XTB calculations. New evaluations update the surrogate models, and adaptive confidence tuning adjusts bootstrap α parameters to maintain uncertainty calibration. This iterative search continues, producing a progressively better Pareto front over multiple objectives like spin-crossover energy, solvation energy, dipole moment, and HOMO-LUMO gap.
Technical innovations
- Application of linear graphlet-based meta-learning surrogates to multi-objective molecular search, enabling fast, interpretable, and data-efficient transfer across objectives.
- Integration of Bayesian bootstrap ensembles for uncertainty quantification in linear surrogate models, capturing distributional mismatch without Gaussian assumptions.
- Development of an adaptive confidence-tuning algorithm that dynamically recalibrates bootstrap concentration parameters to maintain calibrated uncertainty in evolving search distributions.
- Use of functionalization-based candidate sampling combined with K-means clustering within an EGO framework to balance exploration and exploitation over large chemical spaces.
Datasets
- QM9 — approximately 134,000 small organic molecules with DFT properties — public benchmark
- Spin Crossover Metal-Organic Complexes (SCO) — size varies per iteration; ligand pool built from chemically filtered subsets of the Cambridge Structural Database (non-public)
Baselines vs proposed
- Random sampling: Pareto front coverage c-metric ~20% vs meta-learning: >80% (QM9, Fig. 3a)
- Base linear regression surrogate: Pareto front dominated by meta-learning surrogate in 78% of cases (SCO, Fig. 5a)
- Base model RMSE on SCO candidates reduced up to 47% by meta-learning surrogates (no specific figure cited)
- Static uncertainty calibration: Pareto front dominated by adaptive confidence tuning in 52% of cases (SCO, Fig. 5b)
Figures from the paper
Figures are reproduced from the source paper for academic discussion. Original copyright: the paper authors. See arXiv:2606.20497.
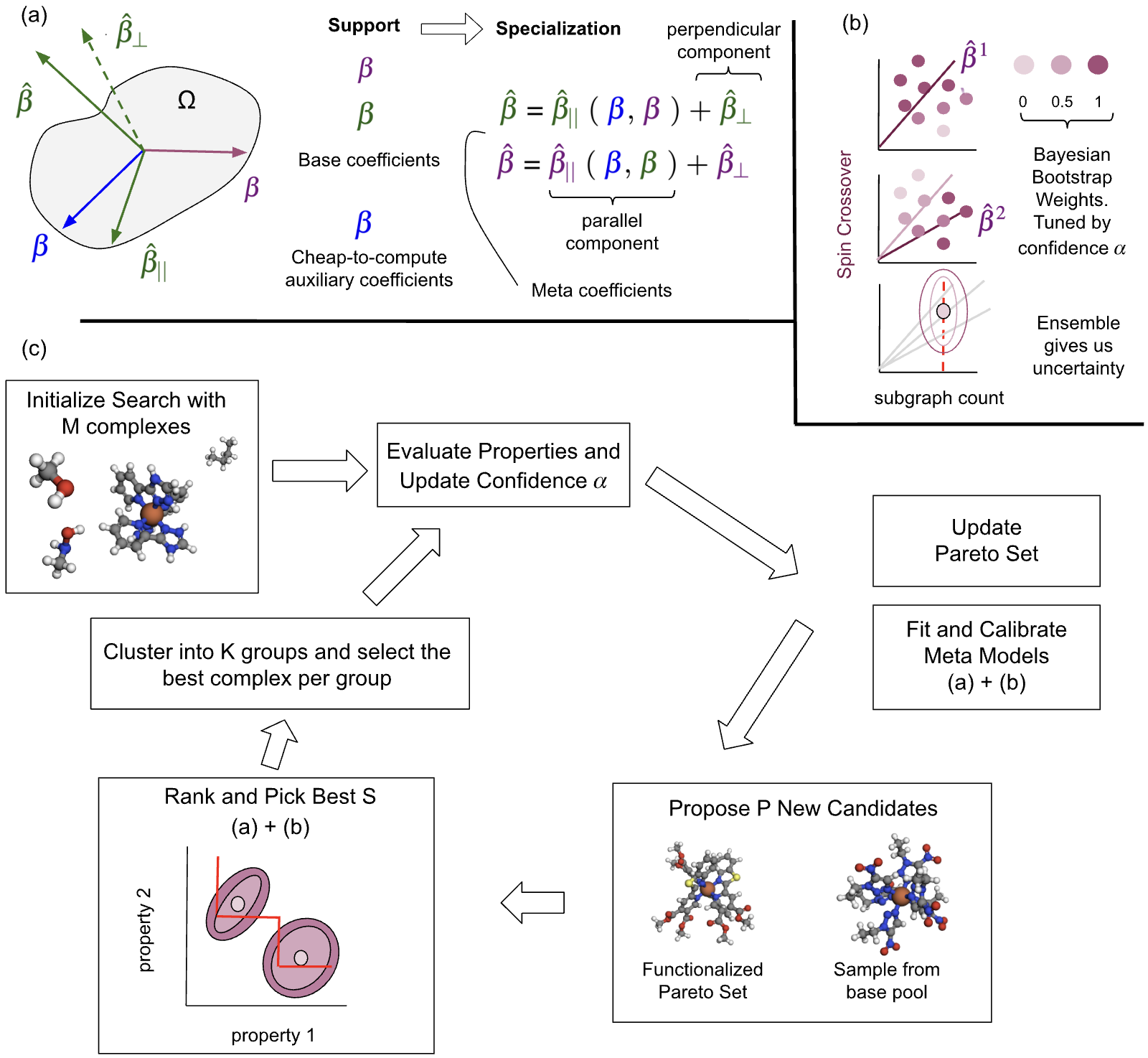
Fig 1: Core components of the proposed multi-objective chemical search framework. (a) The meta-
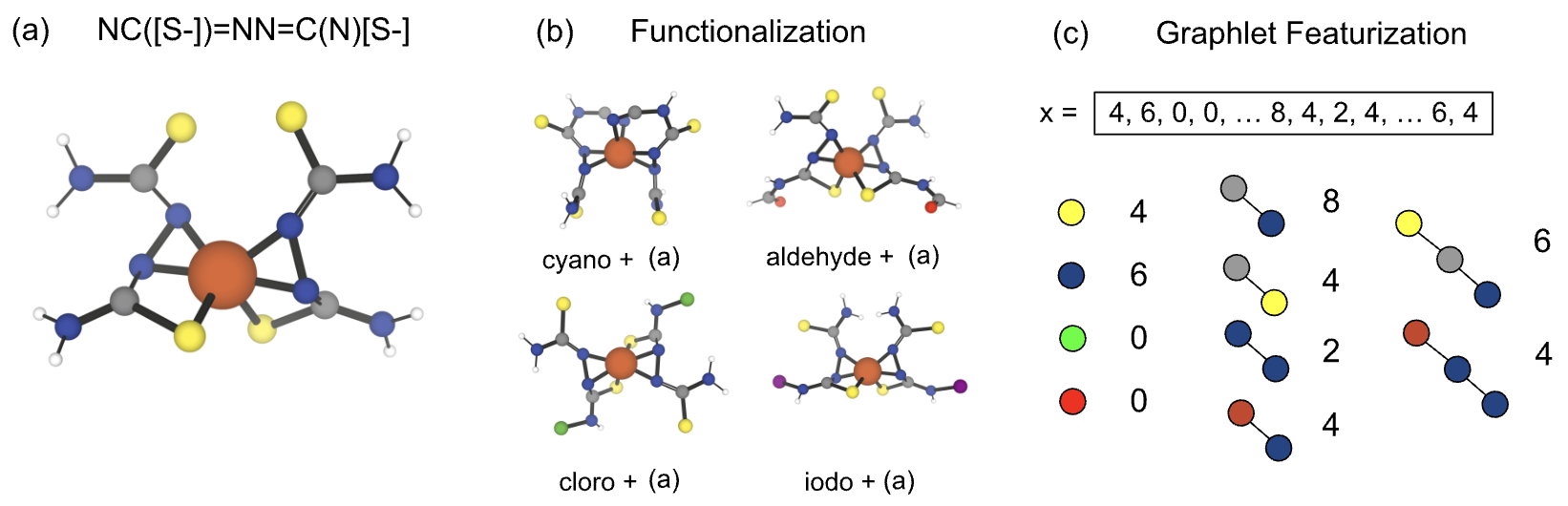
Fig 2: Functionalization-based molecular generation and graphlet featurization. (a) Example complex
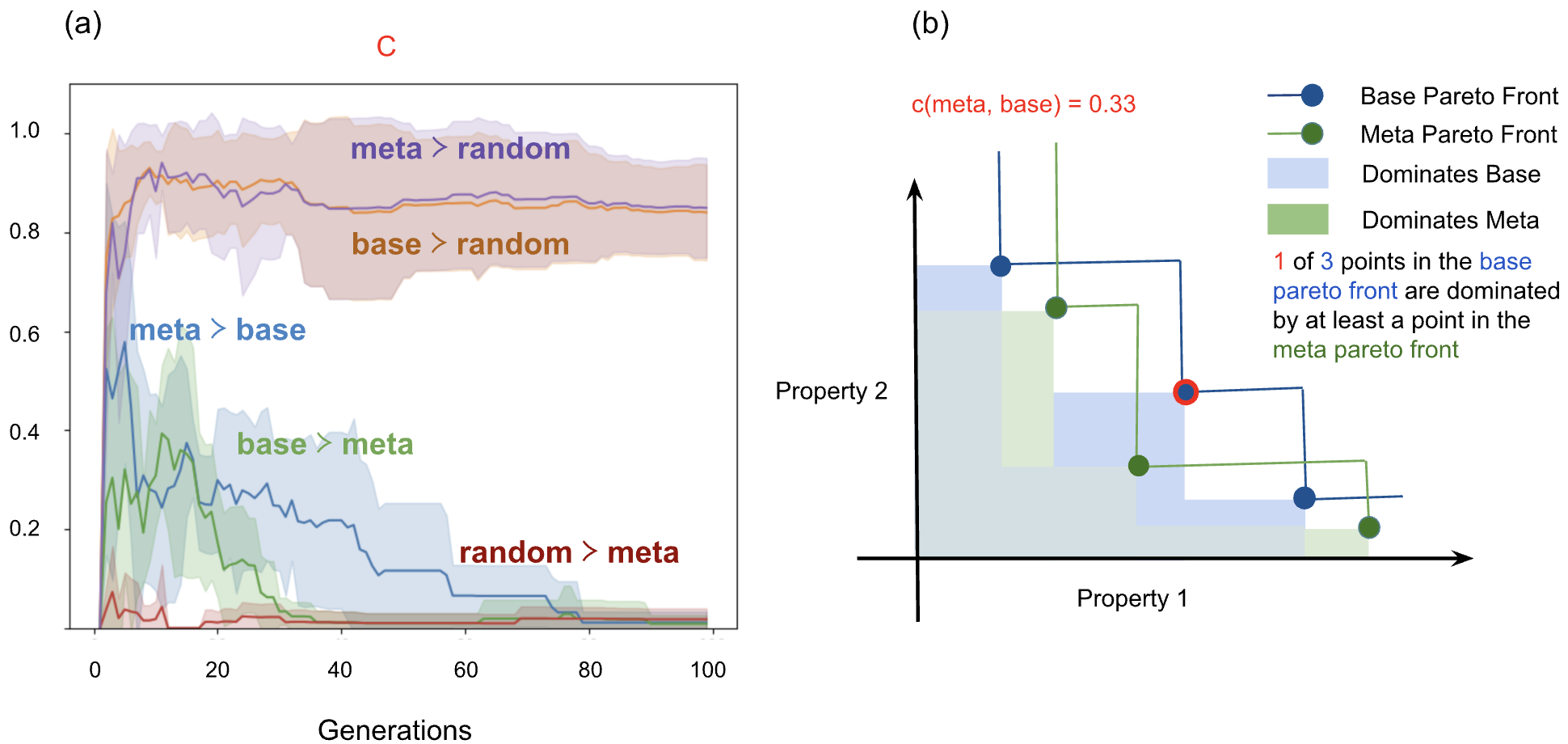
Fig 3: Pareto-front coverage on QM9. (a) C-metric results for the QM9 experiment, averaged over five
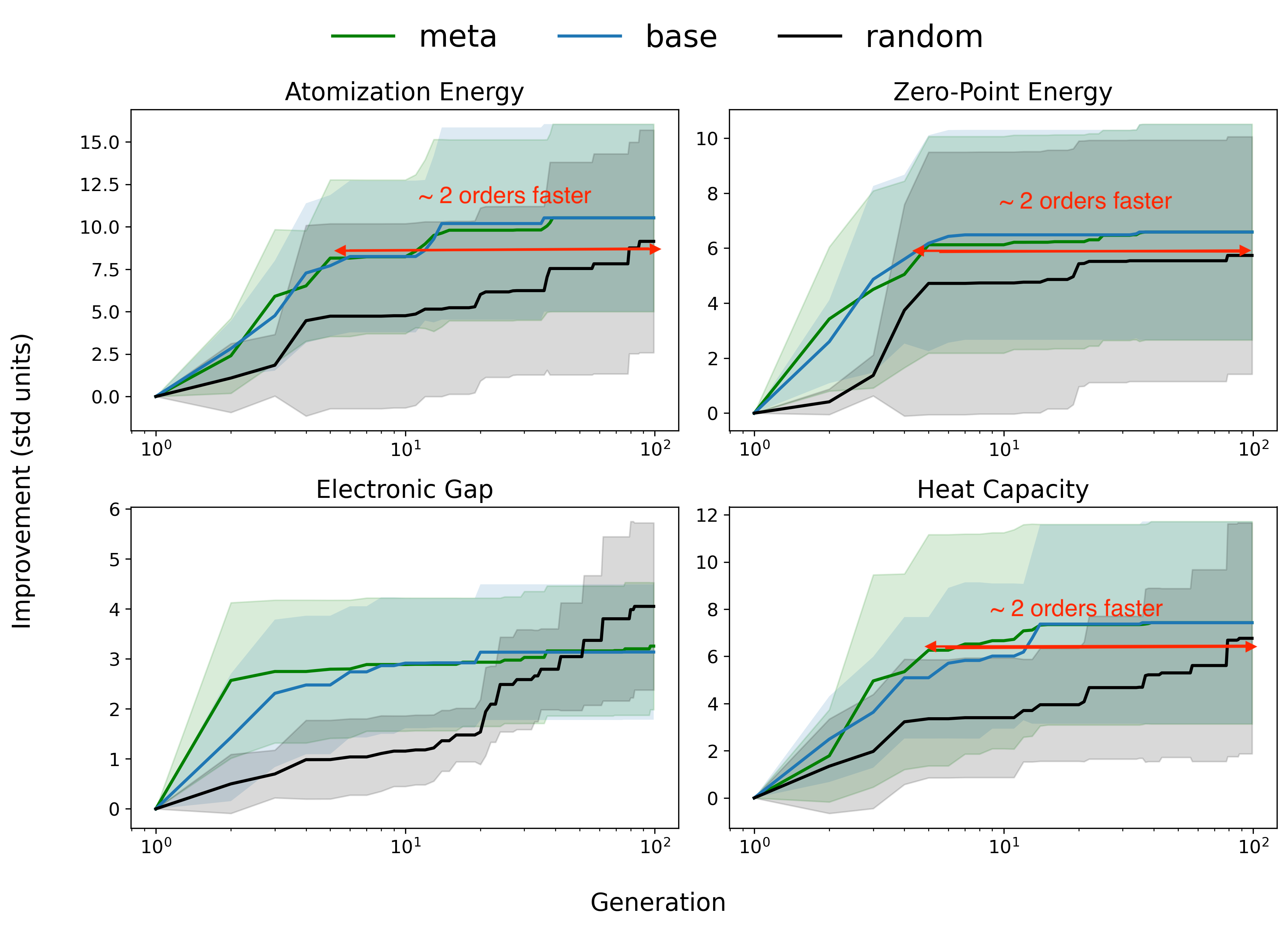
Fig 4: Relative improvement of optima in the marginal distribution of individual QM9 properties.
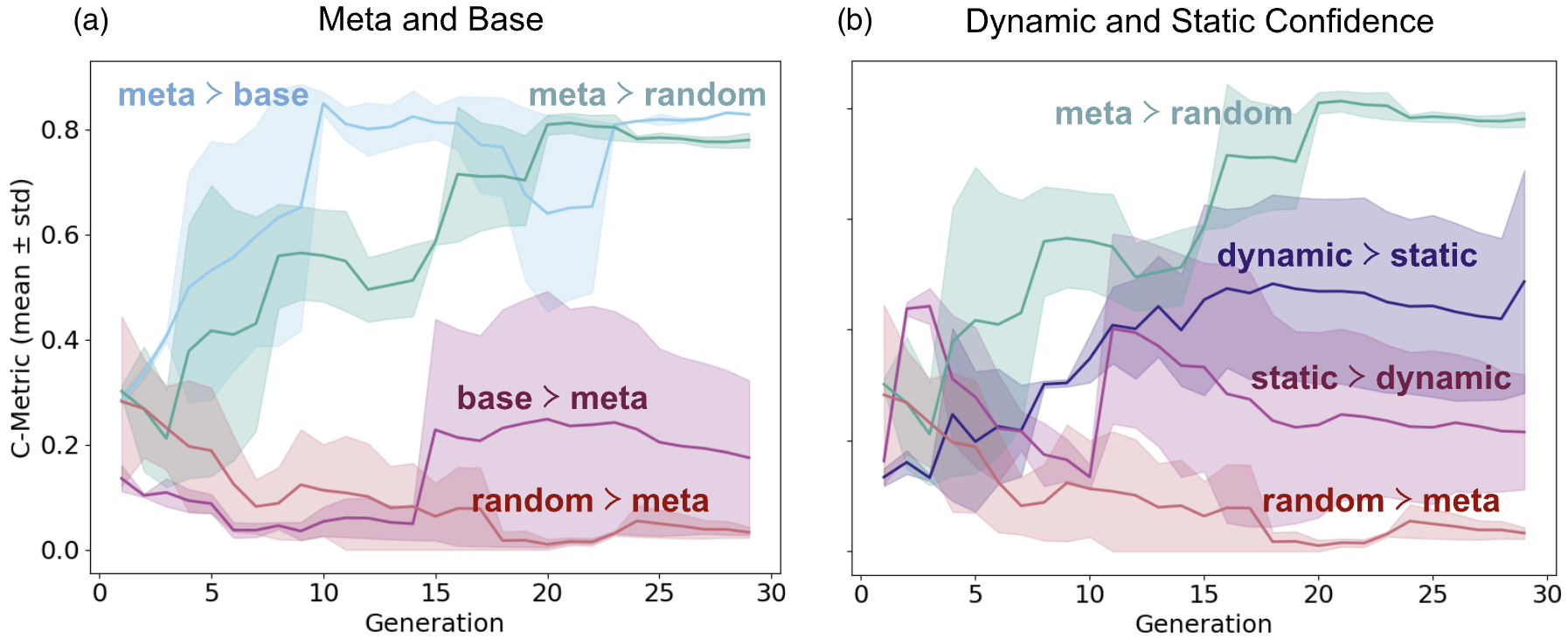
Fig 5: Pareto-front dominance in the spin-crossover search. (a) C-metric values for the spin-crossover
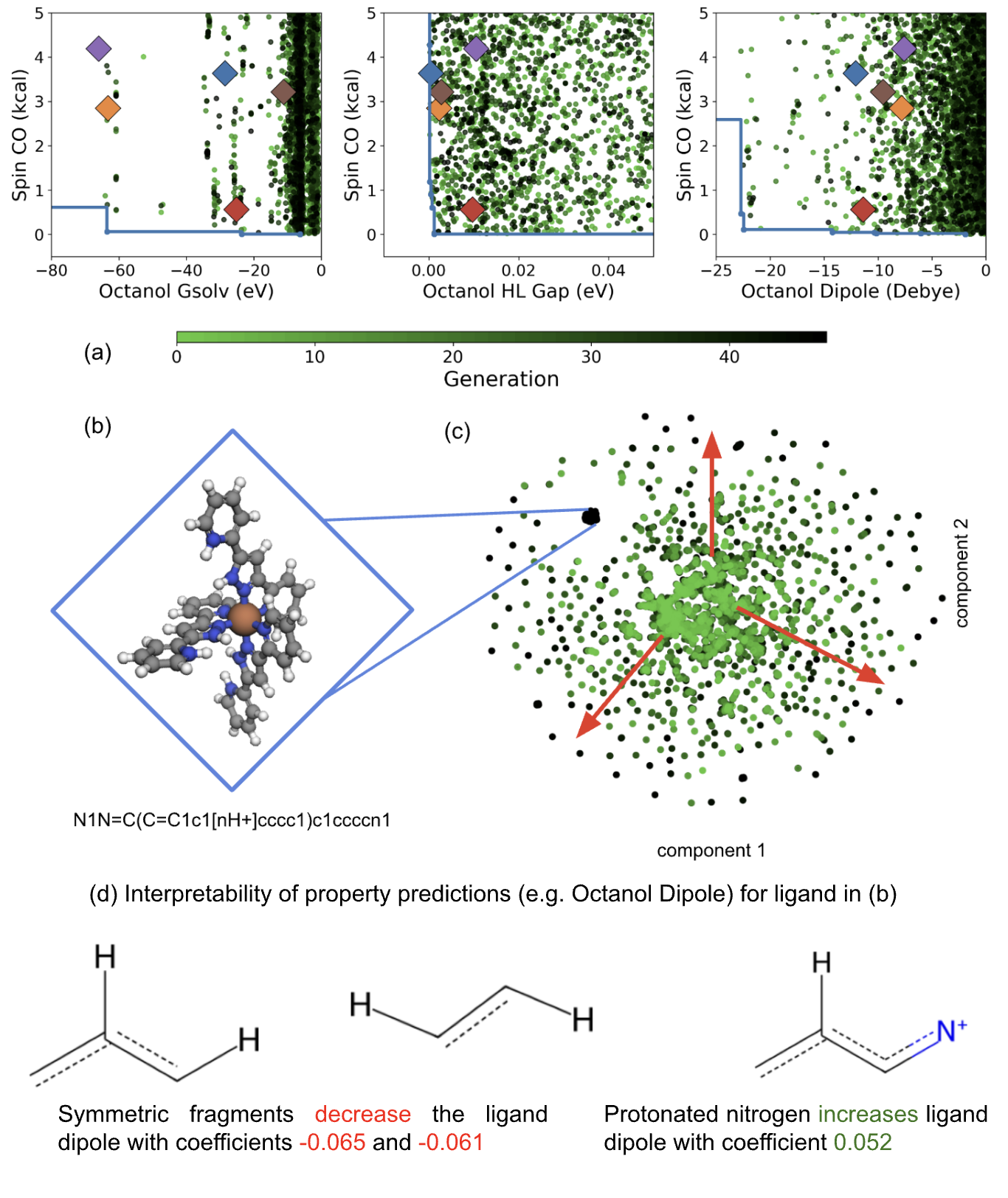
Fig 6: Pareto-optimal complexes, chemical-space exploration, and model interpretability in the SCO
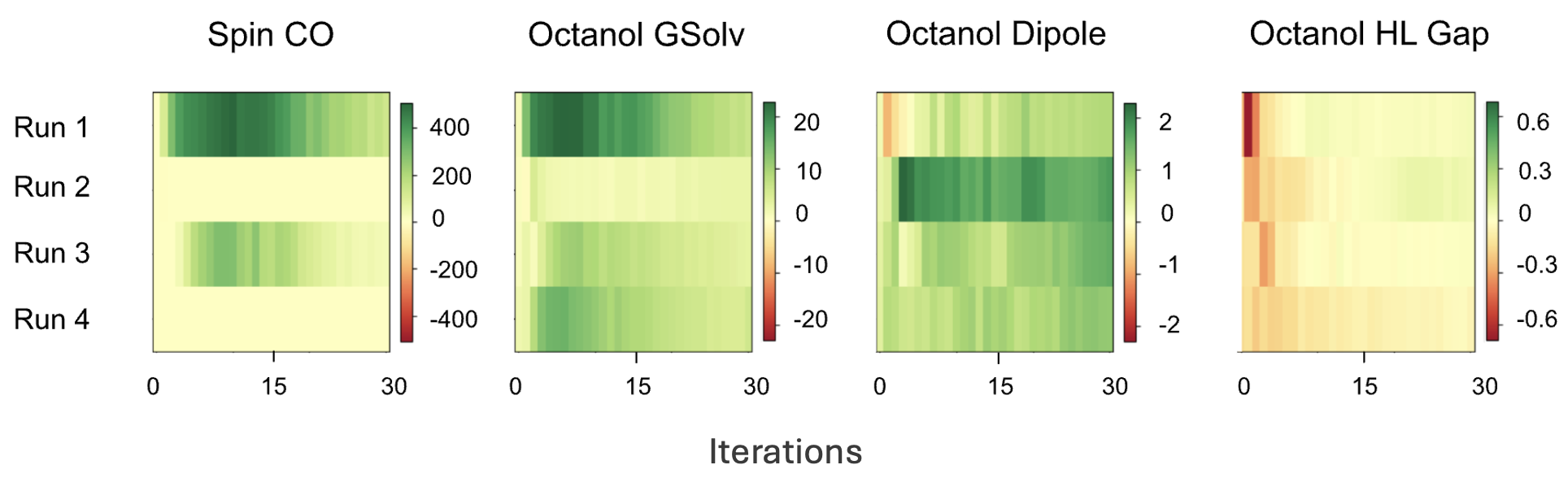
Fig 7: Property prediction error on meta-selected SCO candidates over the course of search. Prediction
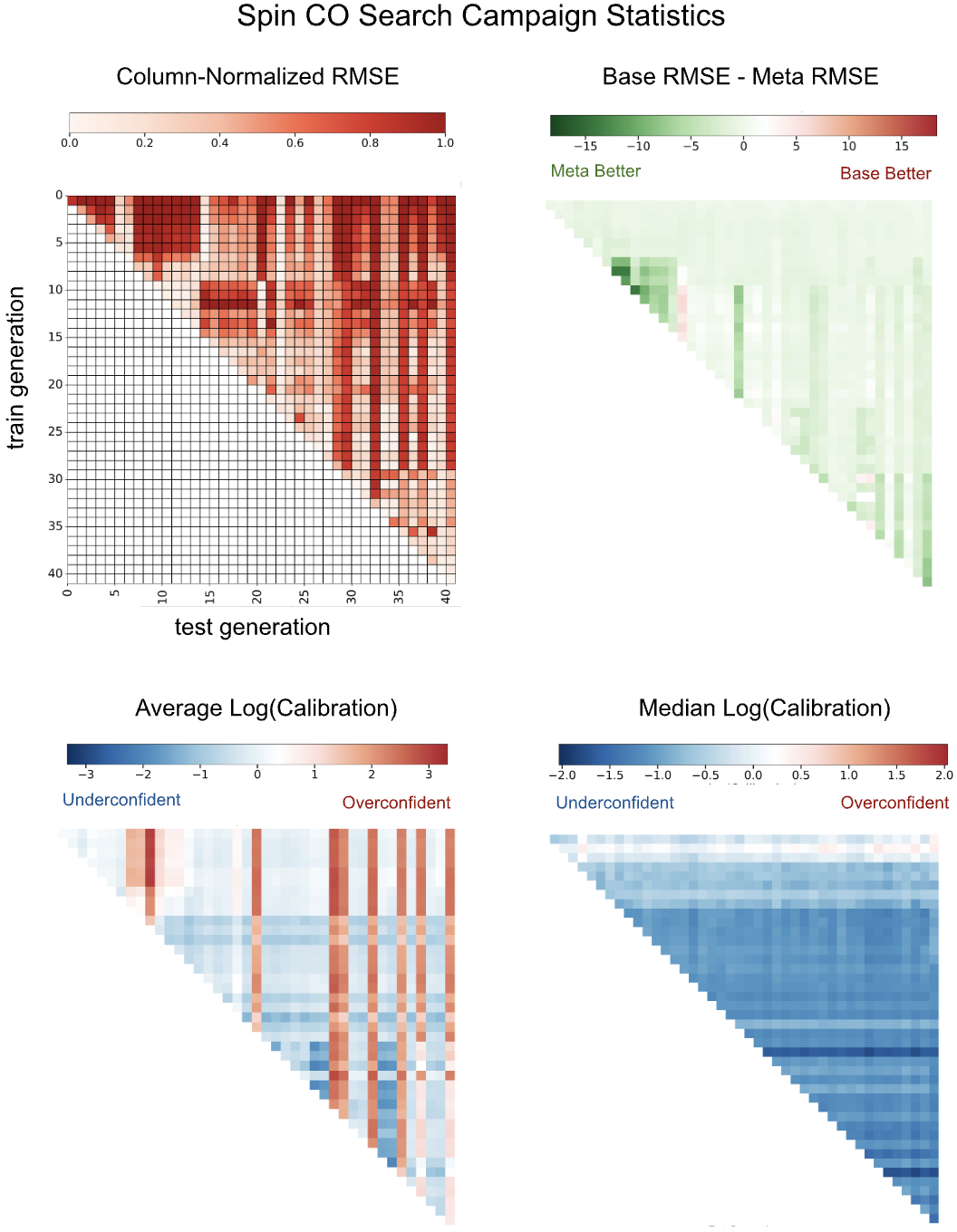
Fig 8: Out-of-distribution robustness and uncertainty calibration for the spin-crossover target across
Limitations
- The SCO search space is extremely large and full convergence was not established within the evaluation budget; results show partial search progress.
- Uncertainty quantification does not fully address out-of-distribution or adversarial molecular candidates, though adapts dynamically.
- The approach is limited to linear surrogates on graphlet features; nonlinear or deep models might capture more complex chemical interactions at increased cost.
- Code release, full reproducibility details, and exact hyperparameters are not explicitly provided.
- The adaptive confidence tuning relies on heuristic log-error measures and confidence update rules whose theoretical guarantees are unclear.
- The study focuses on surrogate model accuracy and multi-objective search performance without exploration of long-term chemical synthesis validation.
Open questions / follow-ons
- How would nonlinear or deep meta-learning surrogates compare to linear graphlet models in multi-objective chemical search in terms of interpretability and efficiency?
- Can the adaptive confidence tuning approach be theoretically grounded or extended to other uncertainty quantification frameworks?
- How robust is the pipeline to true out-of-distribution molecular candidates, e.g., arising from novel substructures unseen in any support tasks?
- Can meta-learning frameworks be extended to integrate synthesis feasibility and experimental feedback in closed-loop chemical discovery?
Why it matters for bot defense
Although not directly related to CAPTCHA or bot defense, this paper offers valuable insights for bot-defense engineers interested in active search strategies under data scarcity and distributional shift. The adaptive uncertainty tuning and meta-learning approach provide a compelling example of balancing exploration-exploitation and uncertainty recalibration in complex, evolving decision spaces, which are analogous to adversarial arms races in bot detection systems. The interpretable linear surrogate approach also highlights how model interpretability can be preserved without sacrificing performance—an important consideration when building transparent, trustworthy ML defenses. Finally, the focus on multi-objective optimization and Pareto front analysis can inspire practitioners to consider trade-offs across detection accuracy, latency, and user experience in practical defenses.
Cite
@article{arxiv2606_20497,
title={ Interpretable Meta-Learning for Multi-Objective Chemical Search },
author={ Antonio Varagnolo and Yulia Pimonova and Michael G. Taylor and Raphaël Pestourie and Nicholas E. Lubbers },
journal={arXiv preprint arXiv:2606.20497},
year={ 2026 },
url={https://arxiv.org/abs/2606.20497}
}