
PyPeakRankR: Reproducible Peak-Level Feature Extraction for Regulatory Element Ranking
Source: arXiv:2606.18179 · Published 2026-06-16 · By Saroja Somasundaram, Nelson J. Johansen, Trygve E. Bakken, Jeremy A. Miller
TL;DR
PyPeakRankR addresses a key gap in regulatory genomics workflows by providing a standardized, reproducible, and extensible tool to extract a diverse set of peak-level features from chromatin accessibility data (e.g., ATAC-seq). While many existing tools focus on specific features or internal data models, PyPeakRankR unifies signal intensities, sequence composition (GC content), evolutionary conservation (PhyloP scores), and statistical signal distribution moments into a single portable TSV feature matrix. This separation of deterministic feature extraction from ranking enables transparent, reproducible benchmarking of peak prioritization strategies. The tool includes both CLI and Python API interfaces and supports cross-genome assembly comparisons via liftOver, processing thousands of peaks in minutes.
The package was validated in the Brain Initiative Cell Census Network (BICCN) challenge where its predecessor ranked among the top 3 of 16 approaches for cell-type specific enhancer prediction. PyPeakRankR also demonstrated direct experimental utility within a recent cross-species enhancer-AAV toolkit pipeline applied to basal ganglia cell types in mouse and macaque. There, composite rankings derived from the features extracted by PyPeakRankR yielded enhancer-AAV tools with over 70% on-target specificity, including top enhancers surpassing 90%. Overall, PyPeakRankR fills an important reproducibility and integration gap in peak prioritization pipelines with immediate utility for enhancer discovery and AAV tool design.
Key findings
- PyPeakRankR consolidates multiple peak-level features — ATAC signal summaries, GC content, PhyloP conservation, distribution moments (kurtosis, skewness, bimodality), and cell-type specificity scores — into a single TSV matrix.
- The package executes feature extraction deterministically, enabling the same input and BigWig signals to produce identical feature tables, separating extraction from ranking.
- In the BICCN Community Challenge, PeakRankR (the predecessor) ranked among the top 3 of 16 methods for cell-type-specific enhancer prediction in mammalian cortex.
- In a cross-species basal ganglia study, PyPeakRankR-supported rankings outperformed conventional fold-change methods, achieving >70% on-target specificity for enhancer-AAV tools, with some enhancers exceeding 90%.
- Feature extraction runs in minutes on thousands of peaks, supporting large-scale experiments with both CLI and Python API interfaces.
- PyPeakRankR supports cross-assembly scoring with liftOver, allowing comparison across species or genome builds.
- The table-first design enables incremental column addition and easy extension to include future sequence model or spatial epigenomic scores without altering peak coordinates.
Threat model
Not a security-focused paper; however, implicitly assumes an honest data generation process where users have access to accurate peak coordinates, BigWig signal files, and sequence references; threat from inconsistent or ad hoc custom code leading to irreproducible or incomparable prioritisations motivates this standardized tool.
Methodology — deep read
The threat model assumes users wish to prioritize candidate regulatory elements (peaks) derived from chromatin accessibility assays like ATAC-seq for functional validation, requiring reproducible, consistent feature extraction pipelines. PyPeakRankR is designed to prevent inconsistent ad hoc feature computations by separating deterministic feature extraction from downstream ranking and benchmarking logic.
Data consists of input peak coordinate files (e.g., BED or TSV format) and associated BigWig signal files representing chromatin accessibility signal intensities across the genome. Additional sequence data (FASTA) is used to compute GC content, and PhyloP conservation scores are sourced from multispecies alignments. No explicit training is involved; rather, PyPeakRankR aggregates these heterogeneous data sources into a unified peak-by-feature table.
The architecture centers around modular subcommands, each appending one or more feature columns to an existing TSV table without modifying peak coordinates. Key functions include init_table (initializes peak table), add_signal (extracts BigWig signal summaries per peak), add_gc (computes GC content), add_phylop (annotates conservation scores), add_moments (computes signal shape statistics such as kurtosis, skewness, bimodality), and rank_by_specificity (computes cell-type specificity scores normalized to [0,1]). The tool leverages pyBigWig for efficient BigWig access, pyfaidx for FASTA handling, and SciPy for statistical moment calculations.
Training regime is not applicable as this is a feature extraction and scoring pipeline, not a learned model. Instead, performance validation was conducted by using feature matrices produced by PyPeakRankR with various ranking strategies and evaluating their ability to predict validated cell-type-specific enhancers.
Evaluation included benchmarking in the BICCN community challenge where PeakRankR ranked top 3 of 16 methods for enhancer prediction. Additionally, the pipeline was embedded in a cross-species enhancer-AAV toolkit, where composite rankings led to >70% on-target specificity in vivo. The evaluation metrics focused on enhancer prediction accuracy and biological specificity rather than traditional machine learning metrics.
PyPeakRankR is open source under MIT license with code, CLI tool, Python API, unit tests, and example datasets available on GitHub and archived on Zenodo. The underlying sequence and conservation data are public, while performance validation relies partially on proprietary challenge datasets and recent published studies. Reproducibility is supported by deterministic feature extraction and documented commands, but some downstream benchmarking datasets are private or semi-public.
Technical innovations
- Unified, portable peak-by-feature matrix output that combines BigWig signal summaries, GC content, PhyloP conservation, and signal distribution moments for regulatory element ranking, unlike fragmented tools that handle only subsets.
- Separation of deterministic feature extraction from ranking logic enabling transparent benchmarking of prioritization methods on fixed upstream data.
- Flexible table-first pipeline design that incrementally appends feature columns without altering peak coordinates, supporting extensibility and reproducibility in large genomics workflows.
- Dual interface with CLI commands and matching Python API functions ensuring integration in both shell pipelines and interactive notebooks without rewriting logic.
- Cross-assembly peak scoring support via liftOver for cross-species and cross-genome build comparisons, addressing a key need in evolutionary regulatory genomics.
Datasets
- BICCN community challenge dataset — thousands of cortical enhancer candidate peaks — proprietary/controlled release
- Cross-species basal ganglia enhancer dataset — mouse and macaque peak sets from ATAC-seq — unpublished, internal to Allen Institute
Baselines vs proposed
- PeakRankR in BICCN challenge: ranked top 3/16 methods for cell-type-specific enhancer prediction
- Conventional fold-change ranking in basal ganglia study: enhancer-AAV tools <70% specificity vs PyPeakRankR composite ranking: >70% on-target specificity, with best enhancers >90%
Figures from the paper
Figures are reproduced from the source paper for academic discussion. Original copyright: the paper authors. See arXiv:2606.18179.
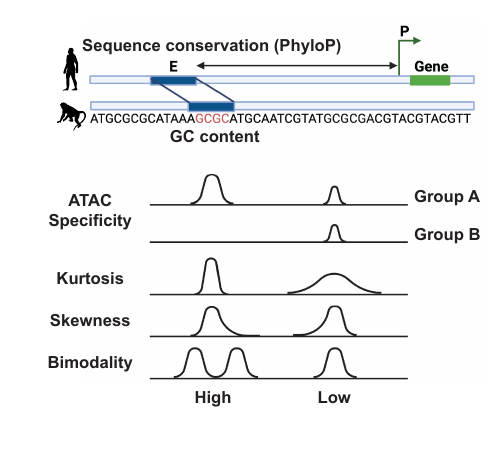
Fig 1: Features collected by PyPeakRankR for each candidate peak: GC content, PhyloP
Limitations
- Dependence on quality and availability of input BigWig and conservation score data; feature accuracy limited by input data quality.
- No adversarial or robustness evaluation against noisy or aberrant peak calls; deterministic extraction assumes consistent input formats.
- Validation concentrated on specific brain cell types and mammalian species (mouse, macaque), unclear generalization to other tissues or organisms.
- Ranking logic provided is relatively simple (ratio-based specificity score); advanced machine learning ranking not integrated but left to user.
- No explicit evaluation under distribution shifts such as rare cell types or varying assay conditions.
- Underlying benchmarking datasets (BICCN, basal ganglia) are partly proprietary, limiting full end-to-end reproduction by external users.
Open questions / follow-ons
- How well do PyPeakRankR features and rankings generalize to non-brain tissues and disease contexts with distinct chromatin landscapes?
- Can advanced machine learning models leverage the unified feature matrix within PyPeakRankR to improve enhancer prediction beyond ratio-based rankings?
- How robust is PyPeakRankR feature extraction to noisy or low-coverage ATAC-seq data or when using novel chromatin accessibility assays?
- Can spatial transcriptomic or epigenomic data (e.g., MERFISH) be effectively integrated into this framework for improved contextual peak prioritization?
Why it matters for bot defense
For bot-defense and CAPTCHA engineering, this paper is tangential but relevant in the broader context of robust, reproducible feature extraction pipelines. PyPeakRankR exemplifies best practices in building deterministic, modular tools that separate core feature computation from downstream decision logic, aiding transparent benchmarking and extensibility. While it operates in the genomics domain, the principles of creating standardized, portable feature matrices and separating deterministic upstream processing from heuristic or learned ranking strategies can inspire more rigorous approaches to feature extraction and scoring in bot activity or human verification challenges.
Bot-defense engineers can also appreciate the importance of reproducibility and benchmarking posed by heterogeneous data, analogous to integrating multiple signals (behavioral, device, timing) into unified representations for robust classification. Techniques such as succinct feature storage, cross-context normalization, and support for multi-modal or cross-domain data (e.g., cross-assembly in PyPeakRankR) provide architectural ideas for designing scalable, extensible detection pipelines beyond genomics.
Cite
@article{arxiv2606_18179,
title={ PyPeakRankR: Reproducible Peak-Level Feature Extraction for Regulatory Element Ranking },
author={ Saroja Somasundaram and Nelson J. Johansen and Trygve E. Bakken and Jeremy A. Miller },
journal={arXiv preprint arXiv:2606.18179},
year={ 2026 },
url={https://arxiv.org/abs/2606.18179}
}