
Flexible Kernels for Protein Property Prediction
Source: arXiv:2606.11057 · Published 2026-06-09 · By Martin Jankowiak, Yerdos Ordabayev, Rudraksh Tuwani, Henry N. Ward, Hunter Nisonoff, James M. McFarland et al.
TL;DR
This paper addresses the critical problem of predicting diverse protein properties, such as binding affinity and thermostability, from limited experimental data. Accurate prediction in these sparse data regimes is highly valuable for protein design but remains challenging. The authors introduce a novel family of Gaussian process (GP) kernels—termed LOCK (Locally Linear Correlation Kernels)—that exploit classical evolutionary substitution matrices (e.g., BLOSUM50) combined with local linearity assumptions to create data-efficient models of protein fitness landscapes. The kernels incorporate learnable exponents on substitution matrices to adapt to different protein property landscapes. Beyond sequence-only kernels, the authors generalize LOCK to structure-conditioned kernels called CLOCK that leverage protein foundation model embeddings to produce position-specific amino acid correlation matrices, enabling multi-task learning across many related protein landscapes.
Comprehensive benchmarking on 21 large protein datasets (including binding affinity, thermostability, and fluorescence) shows that LOCK-GP frequently outperforms strong baselines including deep foundation model embeddings (ESM-2) and prior kernel approaches like Kermut-GP. Notably, LOCK-GP achieves higher Pearson correlations and lower mean absolute errors (e.g., 0.914 Pearson R at 1536 training points averaged across datasets) while requiring far fewer parameters and less compute. The structure-conditioned CLOCK-GP further excels in multi-task learning on 371 thermostability landscapes, decisively beating local supervised methods and matching or exceeding large neural sequence-structure models. These results demonstrate the power of biologically informed, flexible kernel design for reliable, scalable protein property prediction in low-data and multi-task settings.
Key findings
- LOCK-GP achieves a Pearson R of 0.914 and MAE of 0.210 averaged across 21 datasets in cross-validation with 1536 training points, outperforming Kermut-GP (Pearson R 0.888, MAE 0.285) and ESM-2-based models.
- LOCK-GP is 2x to 140x faster in training and inference than foundation model-based methods like Kermut-GP and MLP-ESM2, enabling practical scalability.
- LOCK-GP outperforms baselines on out-of-distribution regimes, including extrapolation and unseen mutation splits (e.g., reduces MAE by 22% relative to the next best model at 512 training points extrapolation).
- The non-linear LOCK kernel kLOCK_nl that incorporates substitution matrices consistently outperforms standard RBF kernels and regularization of kernel exponents improves model fit and stability.
- CLOCK-GP, a structure-conditioned generalization using foundation model embeddings, achieves Spearman R ~0.755 on held-out thermostability landscapes at only 50 training points, outperforming CNN models trained on 700 points.
- The learned position-specific substitution matrices in CLOCK-GP adapt amino acid correlations to local structural context, enabling multi-task learning across hundreds of landscapes.
- LOCK-GP with simple one substitution matrix (e.g., BLOSUM50) and ~210 parameters competes with complex models using millions of parameters and explicit structure.
- Kernel-based models provide superior uncertainty quantification as measured by continuous ranked probability score (CRPS) compared to neural network baselines.
Threat model
n/a (the paper focuses on protein property prediction models rather than adversarial or security threats).
Methodology — deep read
The authors tackle predicting protein scalar properties (e.g., binding affinity, thermostability) from amino acid sequences of fixed length, optionally using reference structures. The core method is Gaussian process (GP) regression with novel sequence kernels designed for data efficiency and interpretability.
Threat Model & Assumptions: The problem assumes data scarcity and the aim is to model protein property landscapes with reliable uncertainty estimates. The adversary notion is not directly applicable, but the method is designed to generalize with few labeled examples and across multiple related landscapes.
Data: The experiments use a curated set of 21 protein property datasets (≥1800 data points each, varying property types) for single-task evaluation, and 371 thermostability landscapes for multi-task learning. Datasets include ProteinGym and other experimental and predicted structures. Splits include standard cross-validation, extrapolation with increasing Hamming distance, and unseen mutations regimes.
Kernel Design: The key technical innovation is the LOCK kernel combining a linear kernel kLOCK_lin and a multiplicative kernel kLOCK_nl based on position-specific amino acid correlation matrices derived from classical substitution matrices (BLOSUM family). These kernels incorporate local linearity (additive approximate model for mutations) and learnable exponents α that modulate substitution matrix entries’ contribution. The combined kernel has the form:
kLOCK(x,y) = σ_1^2 kLOCK_nl(x,y) kLOCK_lin(x,y) + σ_2^2 \tilde{kLOCK_lin}(x,y)
where σ_1, σ_2 are kernel scales fit by marginal likelihood maximization.
LOCK exploits infinite divisibility of BLOSUM matrices, allowing elementwise exponentiation to adapt similarity metrics to the protein landscape.
CLOCK Generalization: To incorporate structure, the CLOCK kernel conditions the substitution matrices on local structure embeddings from protein foundation models (e.g. Chroma). Each position’s correlation matrix C_ell is parameterized as an RBF kernel on learned amino acid embeddings, themselves linear maps of structure embeddings. Parameters (weight matrix W mapping embeddings) are fit by profiling out scale hyperparameters in the GP marginal likelihood.
Training: Kernel hyperparameters including exponents, noise σ_n, and kernel scales are optimized by gradient-based marginal likelihood maximization with weak gamma and regularizing priors to avoid overfitting. Local exponents per position have stronger priors. Training uses subsets of data points; details on epochs and batch sizes are not specified but standard GP techniques with cubic complexity apply.
Evaluation: Models are evaluated using Pearson R, Spearman R, Mean Absolute Error (MAE), Root Mean Squared Error (RMSE), Negative Log Likelihood (NLL), and Continuous Ranked Probability Score (CRPS) averaged across datasets and regimes. Baseline comparisons include existing GP kernels like Kermut-GP (foundation model plus structure), Tanimoto-GP (BLOSUM62 kernel), and non-GP methods including neural networks with ESM-2 embeddings.
Multi-task Evaluation: On 371 thermostability landscapes with AlphaFold2 predicted structures, CLOCK-GP is trained on various numbers of landscapes to evaluate performance on held-out landscapes. It is compared to CNNs trained with sequence and structure embeddings, ridge regression, and hybrids combining CLOCK-GP with CNN zero-shot predictors.
Reproducibility: The authors provide open-source code for LOCK-GP. Some datasets are public (ProteinGym); others have experimental or predicted structures. Details on train/test splits and ablation studies are thoroughly documented.
Concrete Example: For a dataset with 192 training points, LOCK-GP fits kernel hyperparameters including position-dependent exponents α_l on BLOSUM50 correlation matrices and kernel scales σ_1 and σ_2 via marginal likelihood maximization. It then predicts properties for novel sequences by GP posterior mean and variance using the locally linear correlation kernel. This approach outperforms Kermut-GP which relies on expensive foundation model embeddings and additional structure features.
Technical innovations
- Development of the Locally Linear Correlation Kernel (LOCK), which combines evolutionary substitution matrices raised to learnable exponents with local linearity assumptions in a product kernel for Gaussian processes.
- Utilization of the infinite divisibility property of BLOSUM substitution matrices to support learnable exponentiation of matrix elements, allowing landscape-specific adaptation of amino acid similarity encoding.
- Extension to structure-conditioned kernels (CLOCK) by mapping protein foundation model embeddings to position-specific amino acid correlation matrices, enabling multi-task learning over many protein property landscapes.
- A principled hyperparameter optimization approach with priors tailored for kernel exponents and noise scales, improving numerical stability and preventing overfitting in sparse data regimes.
Datasets
- ProteinGym subset — ~1800+ sequences per dataset — public
- 21 curated protein property datasets including thermostability, binding affinity, fluorescence, capsid viability — ≥1800 datapoints each — mixed experimental and predicted structures
- 371 thermostability landscapes from Tsuboyama et al. (2023) — ~1000 sequences per landscape — public with predicted AlphaFold2 structures
Baselines vs proposed
- LOCK-GP: Pearson R = 0.914, MAE = 0.210 (1536 training data points cross-validation) vs Kermut-GP: Pearson R = 0.888, MAE = 0.285
- LOCK-GP: Pearson R = 0.682, MAE = 0.496 (48 training points) vs Tanimoto-GP: Pearson R = 0.517, MAE = 0.588
- LOCK-GP: MAE 22% lower than next best baseline (Tanimoto-GP) in extrapolation regime with 512 training points
- CLOCK-GP Spearman R = 0.755 at 50 training points on multi-task thermostability data vs CNN-EMB-OH Spearman R = 0.498
- LOCK-GP training and inference is 2x – 140x faster than MLP-ESM2 and Kermut-GP
- Ridge-ESM2 and MLP-ESM2 perform worse on unseen mutation sets compared to LOCK-GP and Tanimoto-GP
Figures from the paper
Figures are reproduced from the source paper for academic discussion. Original copyright: the paper authors. See arXiv:2606.11057.

Fig 1: The BLOSUM50 substitution matrix as a correlation

Fig 2 (page 2).
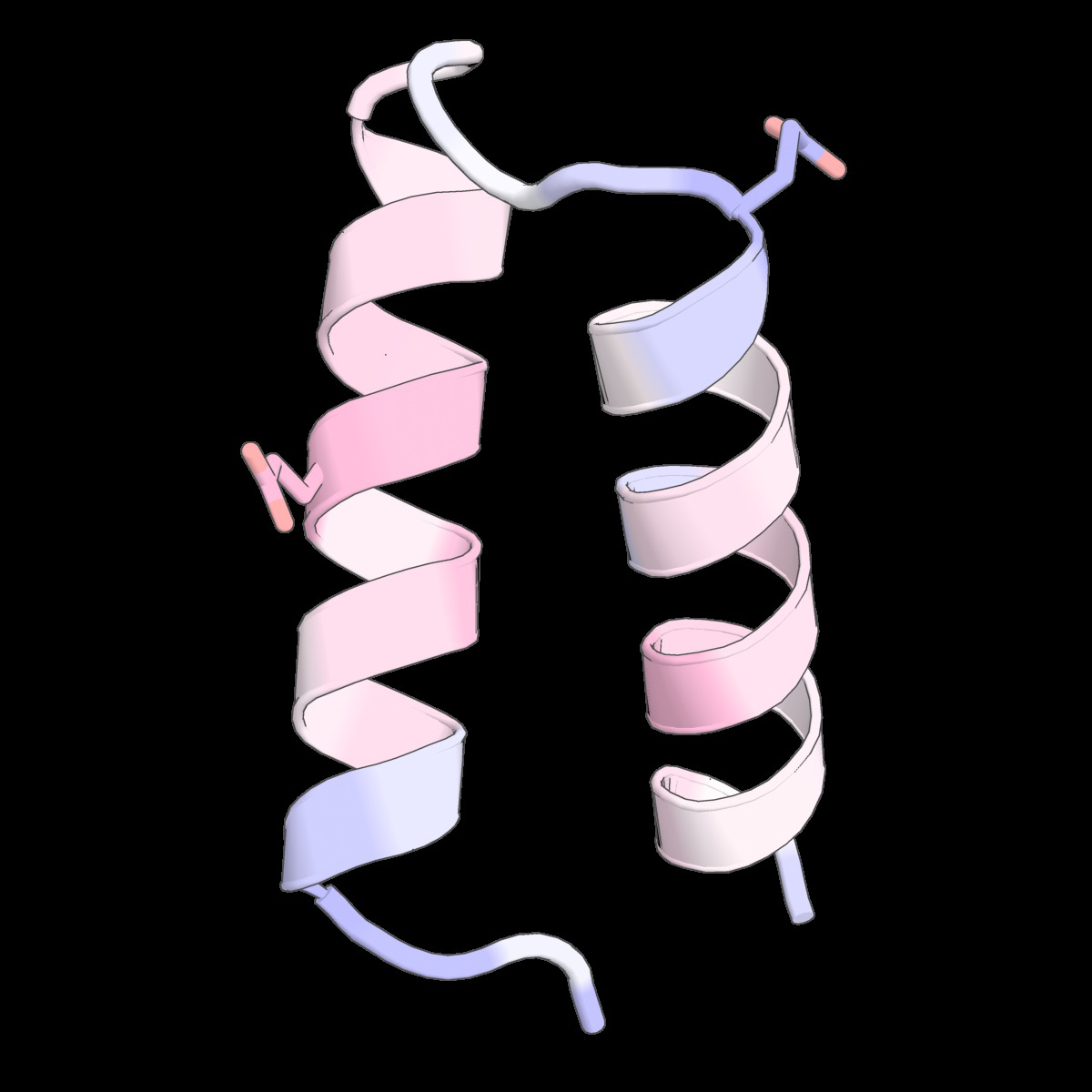
Fig 5: We depict how multi-task model performance changes
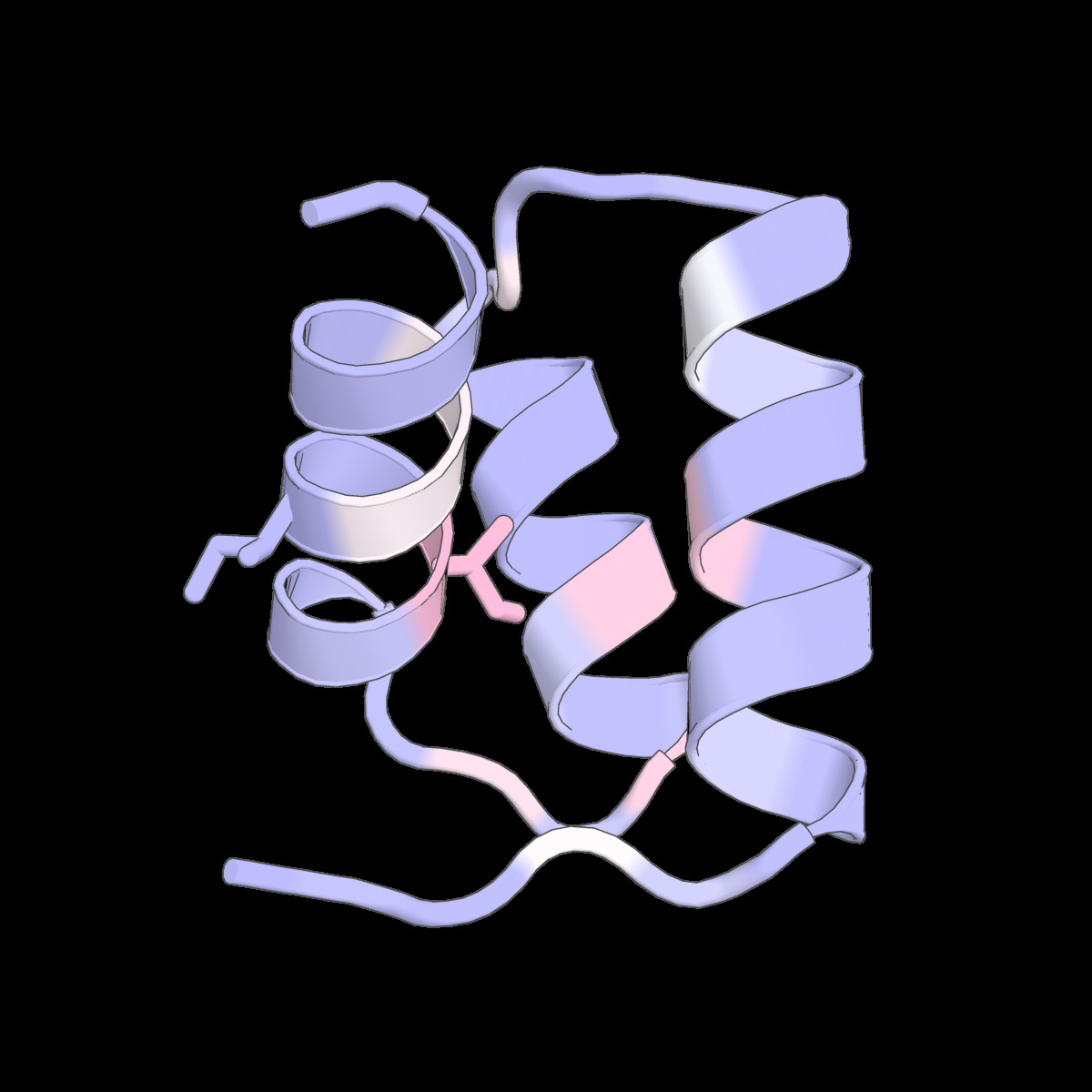
Fig 4: Likewise the Spearman R of CLOCK-GP at N = 50

Fig 6: Structure-conditioned amino acid correlations learned
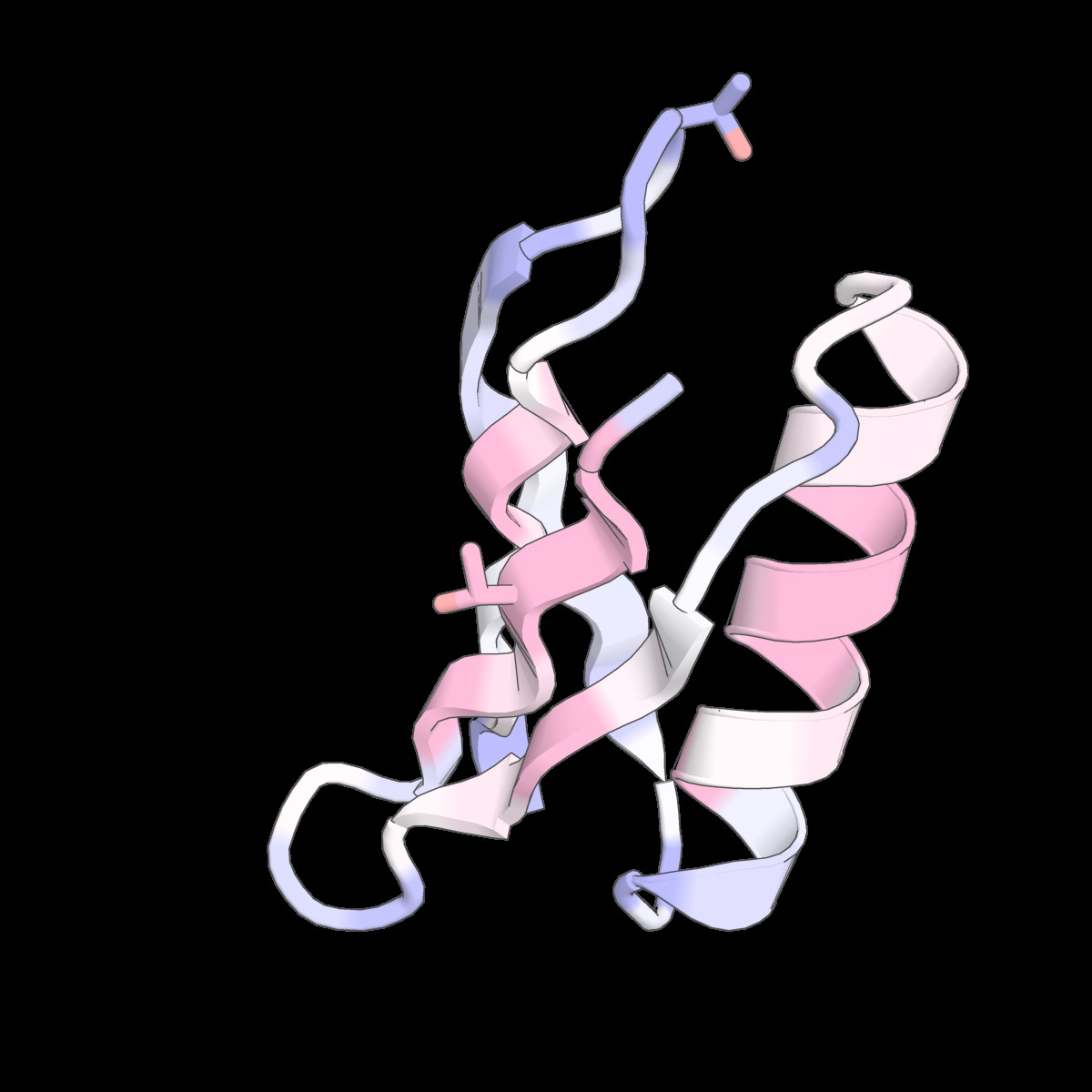
Fig 9: Structure-conditioned amino acid correlations learned by the CLOCK kernel. We show representative structures from Tsuboyama
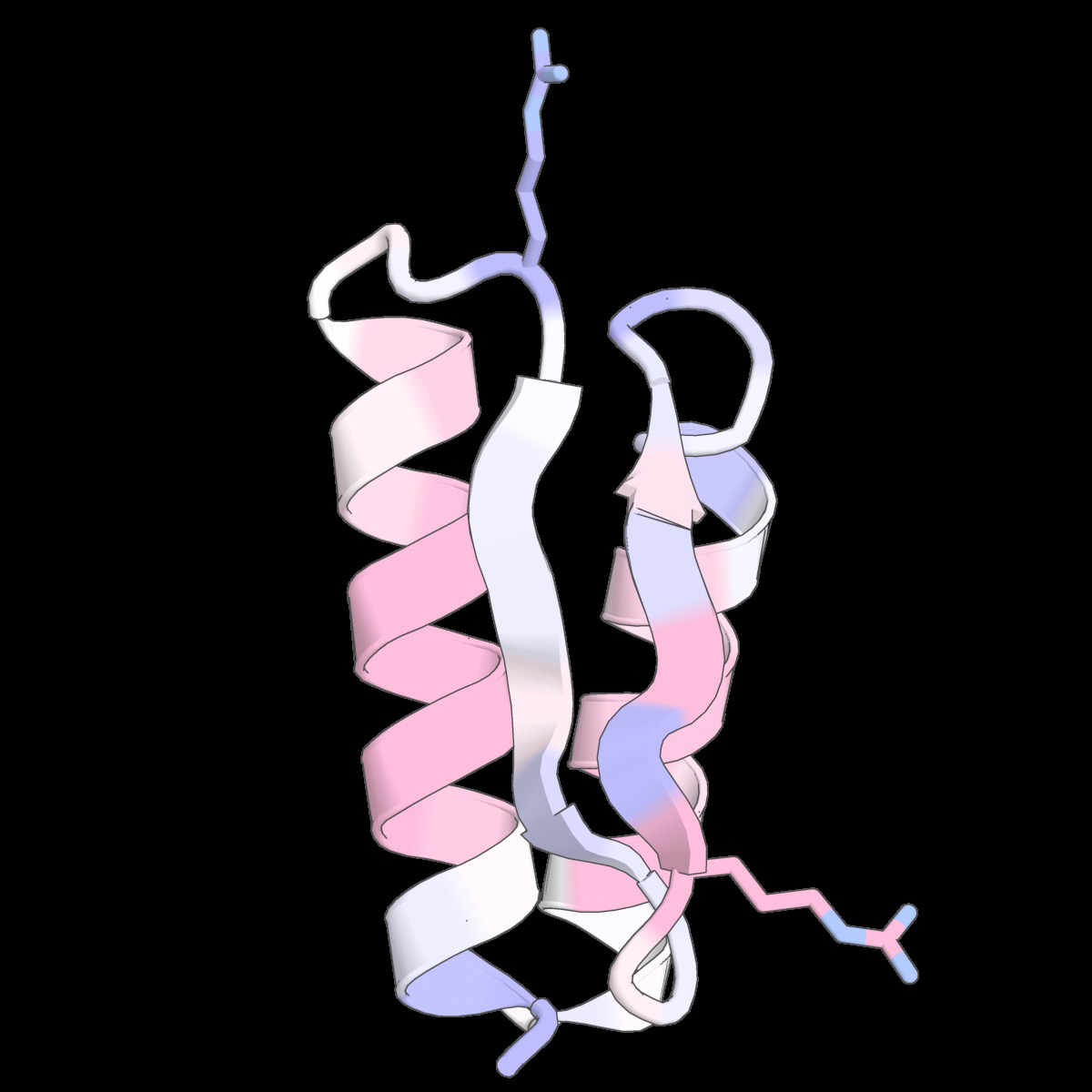
Fig 10: We compare several GPs trained on the AAV dataset from Sinai et al. (2021). We plot both Spearman R (left) and MAE
Fig 11: We compare several GPs trained on the AAV dataset from Sinai et al. (2021). We plot both Pearson R (left) and RMSE
Limitations
- GP training scales cubically with the number of data points potentially limiting applicability to very large datasets, though faster approximations are possible.
- Structure-conditioned CLOCK kernels require sufficiently large multi-task datasets for reliable learning of embedding-to-correlation matrix mappings (e.g., 280+ landscapes).
- The approach focuses on regression of scalar properties and does not directly handle classification tasks or categorical outcomes.
- Evaluation datasets, while large, represent benchmark collections; generalization to very diverse or novel protein families remains to be tested.
- The model does not incorporate explicit higher-order epistasis beyond local linearity and substitution matrix prior knowledge.
- Computational cost and model complexity of foundation models used for structure embeddings remain a bottleneck for some CLOCK variants.
Open questions / follow-ons
- How can the LOCK and CLOCK kernels be extended or scaled to very large protein datasets without incurring cubic computational cost?
- Can these kernels be adapted to explicitly model higher-order epistatic interactions or non-additive effects beyond local linearity?
- What is the robustness of these kernels to distribution shift, e.g., when predicting properties for protein families distant from training data?
- How does the approach perform on protein classification tasks or multi-modal outputs, e.g., predicting binding vs non-binding classes?
Why it matters for bot defense
While not directly related to bot defense or CAPTCHA, this paper exemplifies how carefully designed kernel methods leveraging domain-specific structural priors and evolutionary information can produce highly data-efficient, interpretable models under sparse supervision. Practitioners working on bot detection or CAPTCHA may draw parallels in designing lightweight, uncertainty-aware classifiers that generalize well in low-data regimes by embedding prior domain knowledge (e.g., human interaction patterns) into model kernels or similarity functions. Moreover, the multi-task learning approach to leverage related but distinct data sources efficiently could inspire approaches to aggregate diverse datasets for robust bot detection across different web environments.
Cite
@article{arxiv2606_11057,
title={ Flexible Kernels for Protein Property Prediction },
author={ Martin Jankowiak and Yerdos Ordabayev and Rudraksh Tuwani and Henry N. Ward and Hunter Nisonoff and James M. McFarland and Gevorg Grigoryan },
journal={arXiv preprint arXiv:2606.11057},
year={ 2026 },
url={https://arxiv.org/abs/2606.11057}
}