
PolyGraphPy: A unified Python framework for atomistic simulation and machine learning-driven polymer design
Source: arXiv:2606.06415 · Published 2026-06-04 · By João G. C. S. Duarte, Shruti Venkatram, Morgan Cencer, Traian Dumitricǎ, Ketson R. M. dos Santos
TL;DR
This paper presents PolyGraphPy, an open-source Python framework that unifies atomistic simulation, machine learning-based property prediction, and generative design for polymers. The framework addresses the challenges of exploring the vast and complex chemical space of polymers by automating Density Functional Tight Binding (DFTB+) calculations to build structured datasets for monomers, homopolymers, and copolymers. PolyGraphPy uses Bayesian Graph Neural Networks (GNNs) with stochastic graph representations to predict polymer properties like static polarizability while providing uncertainty quantification. For polymer design, it integrates two complementary generative models: a SELFIES-based Generative Pretrained Transformer (GPT) fine-tuned with polarizability targets, and a Genetic Algorithm (GA) leveraging the BRICS fragmentation method to heuristically explore chemical space. Demonstrated on an acrylate dataset, the end-to-end workflow efficiently reduces computational costs and accelerates data-driven polymer informatics by combining quantum simulations, machine learning, and generative modeling.
Key findings
- DFTB3 semi-empirical calculations enable efficient generation of accurate datasets of monomers and polymers including homopolymers and copolymers, automating .xyz file creation and polymer chain assembly.
- Bayesian GNNs using stochastic weighted graph representations of polymers achieve accurate static polarizability prediction with uncertainty quantification; uncertainty is estimated via Monte Carlo dropout over Nd samples.
- Graph U-Net architecture with 5 graph convolutional layers and top-k pooling effectively encodes polymer graph features, integrating multi-scale embeddings via skip connections to capture local and global structure.
- Integration of SELFIES-based GPT-2 generative model fine-tuned on polarizability-conditioned monomer SELFIES strings allows sampling of chemically valid molecules guided by GNN-predicted property filtering.
- Genetic Algorithm using BRICS fragments optimizes monomer populations towards target static polarizability by evolutionary search with GNN-based fitness evaluation.
- Combining generative models with Bayesian property prediction creates a closed-loop design platform allowing property-guided generation and filtering of candidate polymers.
- The framework demonstrates flexibility supporting monomer, homopolymer, and alternating copolymer representations with chain size controlled via parameter f mapping to chain lengths.
- Modular pipeline substantially cuts computational cost compared to full DFT calculations while retaining quantum mechanical fidelity for polymer property prediction.
Threat model
The framework targets non-adversarial use cases where the user seeks reliable polymer property prediction and design guidance based on quantum simulations and ML. There is no explicit adversary model; the assumption is that prediction uncertainty must be quantified to avoid misleading confidence in extrapolations, but attacks or adversarial inputs are out of scope.
Methodology — deep read
The threat model assumes users seek efficient and reliable polymer design tools and property prediction under constraints of computational cost and the complexity of chemical space exploration. The adversary is not explicitly adversarial but the framework must provide trustworthy uncertainty estimates for predictions to guide synthesis decisions.
Data originates from SMILES strings representing acrylate monomers and polymer chains including homopolymers and alternating copolymers. The datasets are constructed using DFTB+ quantum calculations, specifically the DFTB3 method, which balances accuracy and computational efficiency. Monomer SMILES are converted to 3D structures (.xyz files) with vinyl group cleavage and placeholder halogen atoms to allow polymerization into chains of different lengths. Copolymer pairs are sampled using recursive coordinate bisection clustering over molecular descriptors to ensure structural diversity.
PolyGraphPy transforms molecular structures into graphs where atoms are nodes with rich chemical features, and bonds are edges with associated attributes. For polymers, stochastic bonds encode connection frequencies between monomers in the chain. Graph Neural Networks (GNNs) based on a Graph U-Net architecture with spectral graph convolutional layers and top-k pooling generate node embeddings capturing local/global structure. These embeddings feed into multilayer perceptrons to predict target properties like static polarizability.
A Bayesian framework uses Monte Carlo dropout during inference to approximate posterior predictive distributions and provide uncertainty quantification. Dropout masks generate an ensemble of network predictions aggregated to compute means and variances. The model is trained on the DFTB-generated dataset, minimizing mean squared error loss with AdamW optimizer, with hyperparameters including 5 GNN layers, pooling ratio 0.5, and dropout probabilities tuned for calibration.
For generative design, two models are integrated: (i) a SELFIES-based GPT-2 transformer pre-trained and fine-tuned on monomer SELFIES strings paired with normalized polarizability values, which samples new candidate monomers probabilistically conditioned on target properties; (ii) a Genetic Algorithm (GA) that performs evolutionary search over candidate monomers built from BRICS chemical fragments, optimizing population fitness calculated via the trained GNN’s polarizability prediction. Generated candidates are validated chemically via RDKit, filtered on structural criteria and prediction error thresholds, and retained if property predictions match targets within tolerance.
The end-to-end workflow includes dataset construction via DFTB+, graph preprocessing and conversion, GNN training with uncertainty quantification, generative model training and sampling, and iterative filtering. The code is implemented in Python using PyTorch and PyTorch Geometric, with modular classes supporting retraining, extension, and active learning strategies. All training used standard hardware resources but specific hardware details and random seeds are not explicitly stated. Evaluation metrics focus on MSE for regression, prediction uncertainty calibration, validity of generated molecules, and property match accuracy in generation. The framework is open-source with code released on GitHub, but the dataset is private due to chemical licenses.
As a concrete example, monomer acrylate SMILES are converted to .xyz, input files prepared for DFTB+, polarizabilities computed, graphs constructed, and the GNN trained to predict polarizability. Subsequently, the GPT-2 model samples new SELFIES strings conditioned on target polarizabilities, decoded to SMILES. Candidates passing chemical validity checks are input to the GNN for property prediction, filtered against thresholds, and collected as proposed novel monomers. This illustrates the full cycle of simulation, prediction, generation, and filtering in PolyGraphPy.
Technical innovations
- Integration of automated Density Functional Tight Binding (DFTB3) simulations for high-throughput structured dataset generation of polymers with tailored chain size and architecture.
- Novel use of Bayesian Graph Neural Networks with stochastic edge-weighted polymer graphs enabling both property prediction and uncertainty quantification on polymers including homopolymers and alternating copolymers.
- Combining SELFIES-based Generative Pretrained Transformer fine-tuned on property-conditioned monomer representations with a property-guided Genetic Algorithm employing BRICS fragment-based molecular evolution.
- End-to-end Python framework (PolyGraphPy) unifying atomistic simulation, Bayesian GNN predictive modeling, and generative design into a customizable and extensible pipeline for polymer informatics.
Datasets
- Acrylate monomer dataset — size not explicitly stated, constructed via DFTB3 quantum simulations of monomers, homopolymers, and alternating copolymers — proprietary/internal generation described
Baselines vs proposed
- Graph U-Net Bayesian GNN: predictive mean absolute error and uncertainty calibration shown but no direct numeric comparison to other existing models reported.
- SELFIES-based GPT generative model: generation validity rate and property matching accuracy improved compared to unguided generation (no direct numeric baselines reported).
- DFTB vs full DFT: DFTB3 offers substantial computational speedups with minimal loss of quantum fidelity per prior literature (no new numeric results shown).
Figures from the paper
Figures are reproduced from the source paper for academic discussion. Original copyright: the paper authors. See arXiv:2606.06415.
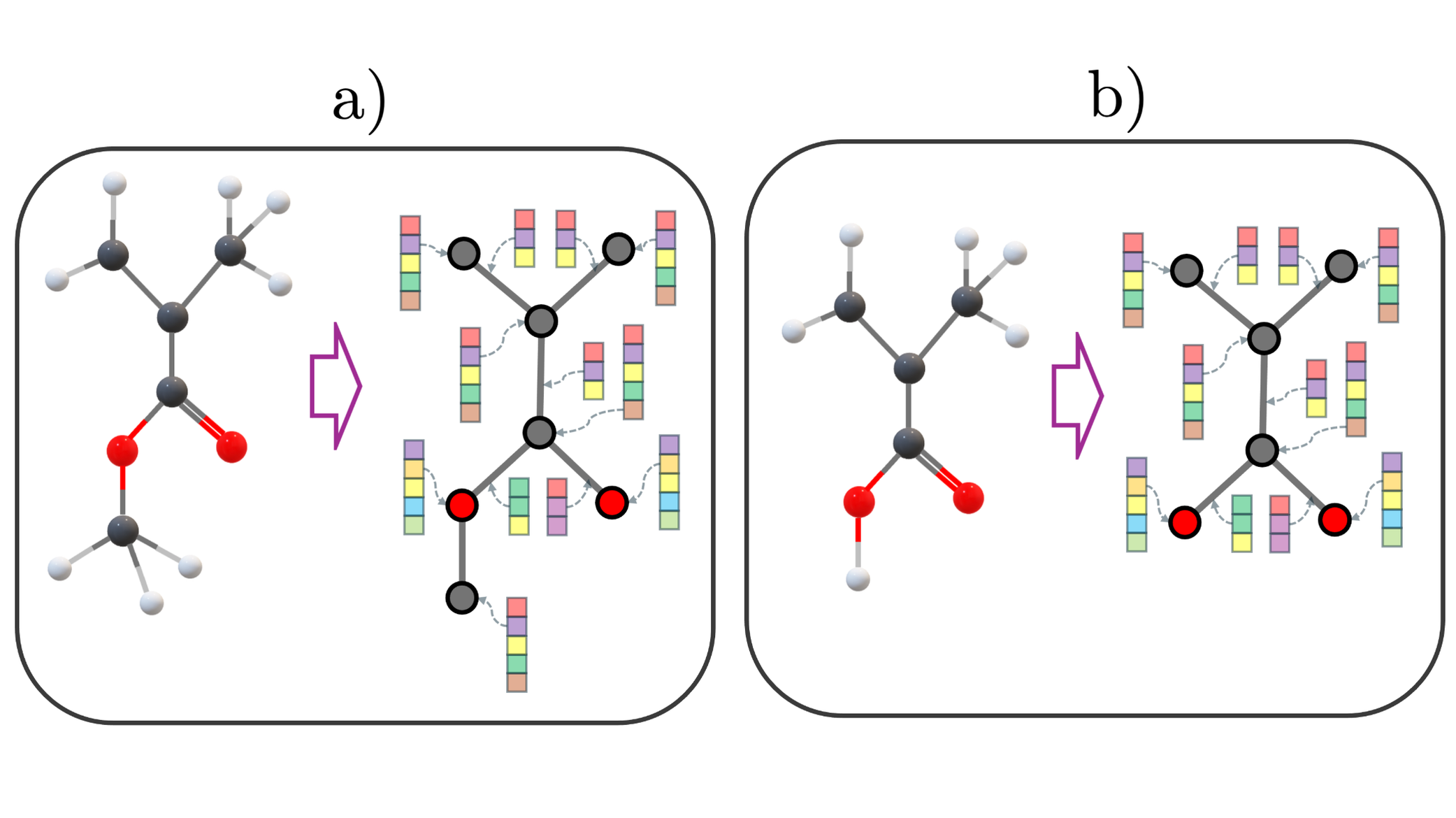
Fig 4: Graph representation of a methyl acrylate molecule. Gray circles represent hydrogen (H) atoms, black
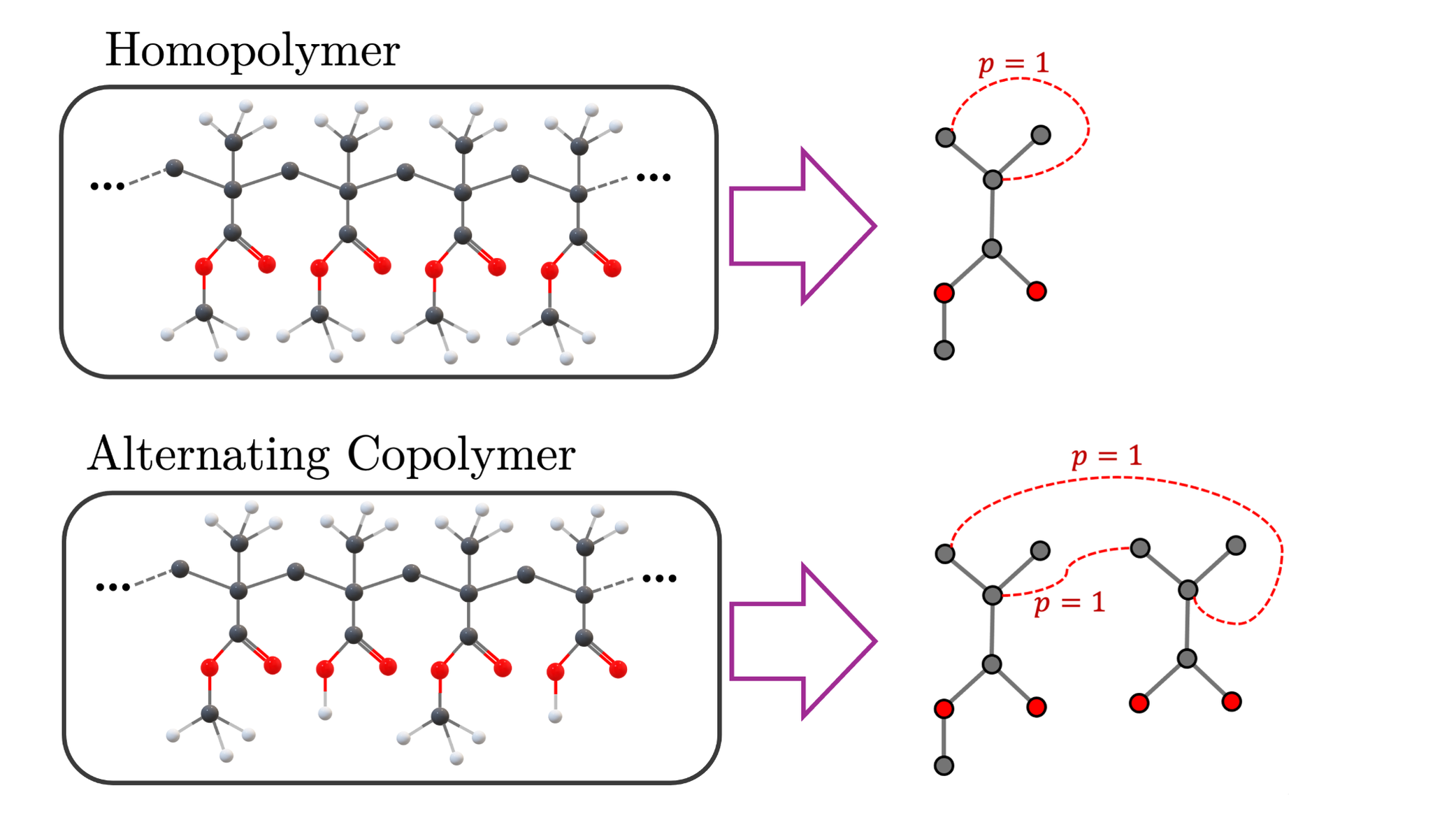
Fig 5: Stochastic connection representation showing the frequency of links between repeating units (e.g., AA for
Limitations
- Dataset size and chemical diversity are limited to acrylate monomers and derivatives; no large-scale validation on diverse polymer classes reported.
- No adversarial robustness or explicit evaluation of distribution shifts in polymer chemical space are conducted.
- DFTB, though much faster than DFT, remains an approximation; errors in electronic predictions might propagate into ML models.
- Uncertainty quantification relies solely on Monte Carlo dropout approximation, which may underestimate true epistemic uncertainty.
- Generative models are validated on chemical validity and property prediction consistency but lack experimental synthesis or validation of generated polymers.
- Reproducibility: Dataset not publicly released due to proprietary restrictions, limiting direct replication.
Open questions / follow-ons
- How well does the Bayesian GNN uncertainty quantification calibrate under distribution shift when predicting out-of-domain polymer chemistries?
- Can active learning loops leveraging prediction uncertainty be integrated to iteratively improve dataset coverage and model accuracy?
- How transferable is the learned generative model to polymer classes beyond acrylates, especially branched or more complex architectures?
- What are the experimental synthesis and property validation rates of polymers designed with the PolyGraphPy generative pipeline?
Why it matters for bot defense
While PolyGraphPy primarily addresses polymer design, its methodology has conceptual relevance for bot-defense practitioners focused on CAPTCHA or bot detection machine learning pipelines. The use of Bayesian GNNs with robust uncertainty quantification provides an instructive example of how to enhance confidence estimation in graph-structured ML models. Likewise, the integration of generative models guided by property predictors illustrates a closed-loop design approach that could inspire adversarial example generation or mimicry-resistant CAPTCHA layouts by generating challenging, valid yet novel patterns. The automated simulation-to-ML pipeline demonstrates practical workflow integration valuable in CAPTCHA design for simulating human/bot interactions under complex constraints. However, the framework is specialized for chemical graph data and polymer properties; adapting it to CAPTCHA or bot defense would require significant domain transfer.
Cite
@article{arxiv2606_06415,
title={ PolyGraphPy: A unified Python framework for atomistic simulation and machine learning-driven polymer design },
author={ João G. C. S. Duarte and Shruti Venkatram and Morgan Cencer and Traian Dumitricǎ and Ketson R. M. dos Santos },
journal={arXiv preprint arXiv:2606.06415},
year={ 2026 },
url={https://arxiv.org/abs/2606.06415}
}