
Effective Biological Representation Learning by Masking Gene Expression
Source: arXiv:2605.31562 · Published 2026-05-29 · By Kian Kenyon-Dean, Alina Selega, Ihab Bendidi, Jordan M. Sorokin, Luca Bertinetto, David Errington et al.
TL;DR
This work addresses the challenge of learning effective deep representations from high-dimensional, noisy, and batch-affected RNA sequencing (RNA-seq) gene expression data. Despite large transcriptomic foundation models (FMs) existing, many fail to outperform simple linear baselines such as PCA, suggesting current approaches may not extract richer biological signal beyond raw counts. The authors propose TxFM, a novel masked autoencoder (MAE) tailored for RNA-seq count data, combining an asymmetric transformer encoder and MLP decoder trained with a specialized Poisson loss and novel rectified tanh activation. TxFM is trained on DiverseRNA-1.4M, a carefully curated 1.4 million sample dataset spanning single-cell and bulk RNA-seq with rich perturbational diversity.
Through extensive inductive evaluation on held-out genetic perturbation datasets, TxFM substantially outperforms existing large-scale FMs trained on datasets over 100× larger, especially in capturing biologically meaningful cell embeddings and intrinsic gene relationships. Ablations identify key architectural and data curation choices—high (∼90%) masking ratio, Poisson loss, careful count preprocessing, and rectified tanh activation—that enable transfer learning improvements beyond normalized input counts. Gene embeddings learned by TxFM reveal strong recall of known functional gene relationships, supporting novel biological discovery. Overall, this study demonstrates that refined self-supervised masked reconstruction with curated data can unlock transcriptomics representation learning beyond prior art.
Key findings
- TxFM-B trained on DiverseRNA-1.4M (1.4M samples) achieves overall perturbation representation score 39.11 vs 35.62 for same model on atlas-scale TF-Sapiens (57M samples), indicating curation beats scale alone (Table 2).
- TxFM-B outperforms 16 existing FMs including STATE-SE and Tahoe-x1 across held-out RPE1, HEPG2, Jurkat cell lines, despite training on >100x less data and fewer parameters.
- TxFM’s decoder gene parameters recall 42.7% of 155,390 known gene-gene relationships at 95th percentile cosine similarity threshold, beating scVI (40.4%) and all atlas-scale model codebooks (Table 4).
- Poisson reconstruction loss combined with library-size normalization and rectified tanh activation yields significantly better perturbation and gene representation quality than alternatives like NB, ZINB, MSE, or SmoothL1 (Table 5a,b,f).
- Uniform random masking with ~90% mask ratio (2048 genes unmasked) yields better transfer performance than frequency-weighted masking favoring rare or frequent genes (Table 5c,d).
- Increasing encoder size from Small to Base improves performance, but no gain moving to Large model on DiverseRNA-1.4M dataset (Table 5g).
- Inclusion of curated phenoprint-filtered K562 cells and bulk RNA-seq samples jointly improves model transfer quality; removing bulk or perturbed K562 cells reduces performance equivalently (Table 5h).
- PCA postprocessing of gene embedding spaces consistently improves recall of functional gene relationships by 10-40%, indicating biological relationships lie on lower-rank manifolds (Figure 2).
Threat model
The threat model involves generalization to unseen cell lines and genetic perturbations represented by transcriptomic profiles, aiming to isolate meaningful biological signals from noise and batch effects. The adversary is natural variability and batch confounding rather than an active malicious actor. The model cannot rely on external biological priors or annotated gene sets, and must perform inductive transfer to new unseen data distributions.
Methodology — deep read
The authors design TxFM, a masked autoencoder adapted for transcriptomic RNA-seq count data. The threat model focuses on generalizing to unseen cell types and perturbations, using self-supervised learning without external biological priors or protein embeddings. The adversary is unlabeled distributional variation, but no malicious attack considered.
Training data is a newly curated diverse corpus DiverseRNA-1.4M containing approximately 1.4 million bulk and single-cell RNA-seq samples from oncology and perturbation studies, including glioblastoma, K562 CRISPRi perturbations, cancer cell lines, gastric metaplasia, tumor microenvironment, breast cancer, TCGA bulk data, and GTEX bulk RNA-seq. Genes expressed in fewer than 1,000 cells and cells expressing fewer than 2,000 genes are filtered, resulting in a union of 44,349 genes across datasets. A phenoprint curation filters noisy perturbations in K562 data.
Input gene counts x are normalized by library size to 10^5 total counts and log1p transformed. Masked autoencoding randomly hides a large portion of genes (mask ~75-90%) per sample, revealing only a subset K=2048 genes to the model encoder. Unlike typical language models treating sequences, TxFM treats gene expression as an unordered set.
The encoder is a transformer taking concatenated learnable per-gene embeddings Ei and normalized expression values xi as input tokens, along with a CLS token. The transformer produces a contextualized CLS embedding representing the cell. The decoder is a shallow 4-layer MLP that takes this CLS embedding and produces a reconstruction of the entire expression vector.
To respect the count-based data, a novel rectified tanh activation is applied to decoder logits to constrain outputs to valid ranges and stabilize gradients. The loss is a Poisson likelihood reconstruction loss evaluated on the log-normalized expression data. This loss is preferred over negative binomial variants due to better gradient behavior especially on low-to-moderate expression genes.
Training uses Base transformer architecture with 768 hidden dimensions and 159M parameters, over 200 epochs, requiring ~1000 A100 GPU hours. Random uniform masking at 2048 genes per sample is used. Ablations explore other masking ratios and weighted sampling. Training optimizes the Poisson loss with Adam optimizer.
For evaluation, the model is tested inductively on held-out genetic perturbation datasets (RPE1, HEPG2, Jurkat cell lines with thousands of CRISPRi edits) unseen during training. Six metrics measure perturbation representation quality, including linear probing, KNN, batch correction scores, and biological relationship recall. Baselines include raw input, PCA, scVI, and 16 published large-scale FMs trained on tens to hundreds of millions of samples.
Gene relationship recall is computed by measuring cosine similarity in learned gene embeddings against 155,390 known protein-protein and functional interactions, before and after PCA dimensionality reduction.
Ablation studies systematically vary loss functions (Poisson, NB, ZINB, MSE), preprocessing, masking strategies, decoder depth and activations, encoder size, and training data curation, measuring impact on perturbation consistency and gene-gene recall metrics.
Code and pretrained TxFM checkpoints are planned for public release. Datasets used are publicly accessible but integrated and curated by the authors.
Example end-to-end: Given a K562 CRISPRi single-cell gene expression vector (after filtering and library size normalization), 2048 genes are randomly unmasked and embedded with learnable gene tokens concatenated with log1p counts. The transformer encoder processes these tokens plus the CLS token to output a contextualized CLS embedding vector representing the cell's state. The decoder MLP takes this embedding and predicts the full gene expression vector. The Poisson loss between predicted and true normalized log-counts trains the model to reconstruct masked gene expression values. Over training epochs, the model learns gene embeddings and cell representations encoding perturbational biology, enabling transfer to held-out cell lines and functional gene relationship discovery.
Technical innovations
- Application of masked autoencoder architecture adapted with Poisson reconstruction loss and a novel rectified tanh activation tailored for non-negative, count-based RNA-seq data.
- Curated DiverseRNA-1.4M transcriptomic dataset combining single-cell and bulk RNA-seq samples with phenoprint filtering to enhance biological signal quality, enabling superior models trained on fewer samples versus larger atlases.
- Use of a CLS-token-based transformer encoder concatenated with per-gene embeddings and log-normalized counts, followed by a shallow MLP decoder, diverging from token-wise decoders common in language MAEs.
- Demonstration that uniform random masking at ~90% yields better transfer learning in transcriptomics than frequency-weighted masking by gene sparsity.
- Discovery that intrinsic gene relationship signals emerge on low-dimensional manifolds in learned gene parameter spaces, recoverable by PCA-reduction and simple similarity, suggesting new avenues for biological interpretation.
Datasets
- DiverseRNA-1.4M — 1.4 million samples — curated public mixture of bulk and single-cell RNA-seq from oncology and perturbation studies
- GTEX — 10,526 bulk samples — public
- TCGA — 23,733 bulk samples — public
- Glioblastoma (Ruiz-Moreno et al., 2022) — 504,929 single-cell samples — public
- K562 CRISPRi (Replogle et al., 2022) — 502,080 single-cell samples — public
- MixSeq cancer cell lines (McFarland et al., 2020) — 102,205 single-cell samples — public
- sci-Plex compounds (Srivatsan et al., 2020) — 99,300 single-cell samples — public
- Gastric metaplasia (Nowicki-Osuch et al., 2023) — 88,399 single-cell samples — public
- Tumor microenvironment (Guimarães et al., 2024) — 71,585 single-cell samples — public
- Breast cancer (Wu et al., 2021) — 31,542 single-cell samples — public
- TF-Sapiens — 57 million single-cell samples — public atlas-scale corpus
Baselines vs proposed
- Raw normalized counts: overall perturbation score = 35.27 vs TxFM-B (DiverseRNA-1.4M) = 39.11
- STATE-SE (170M samples): overall perturbation score = 36.43 vs TxFM-B (DiverseRNA-1.4M) = 39.11
- scVI-L (1.4M samples): overall perturbation score = 32.85 vs TxFM-B (DiverseRNA-1.4M) = 39.11
- PCA (1.4M samples): overall perturbation score = 33.54 vs TxFM-B (DiverseRNA-1.4M) = 39.11
- Geneformer-v2 (104M samples) gene relationship recall (codebook): 20.2% vs TxFM-B (DiverseRNA-1.4M) decoder: 42.7%
- scGPT (33M samples) gene recall: 31.8% vs TxFM-B (DiverseRNA-1.4M) decoder: 42.7%
- TxFM-B on TF-Sapiens (57M) overall perturbation score = 35.62 vs TxFM-B on DiverseRNA-1.4M (1.4M) = 39.11
- Ablation Poisson loss perturbation score = 37.3 vs MSE loss = 27.2
Figures from the paper
Figures are reproduced from the source paper for academic discussion. Original copyright: the paper authors. See arXiv:2605.31562.
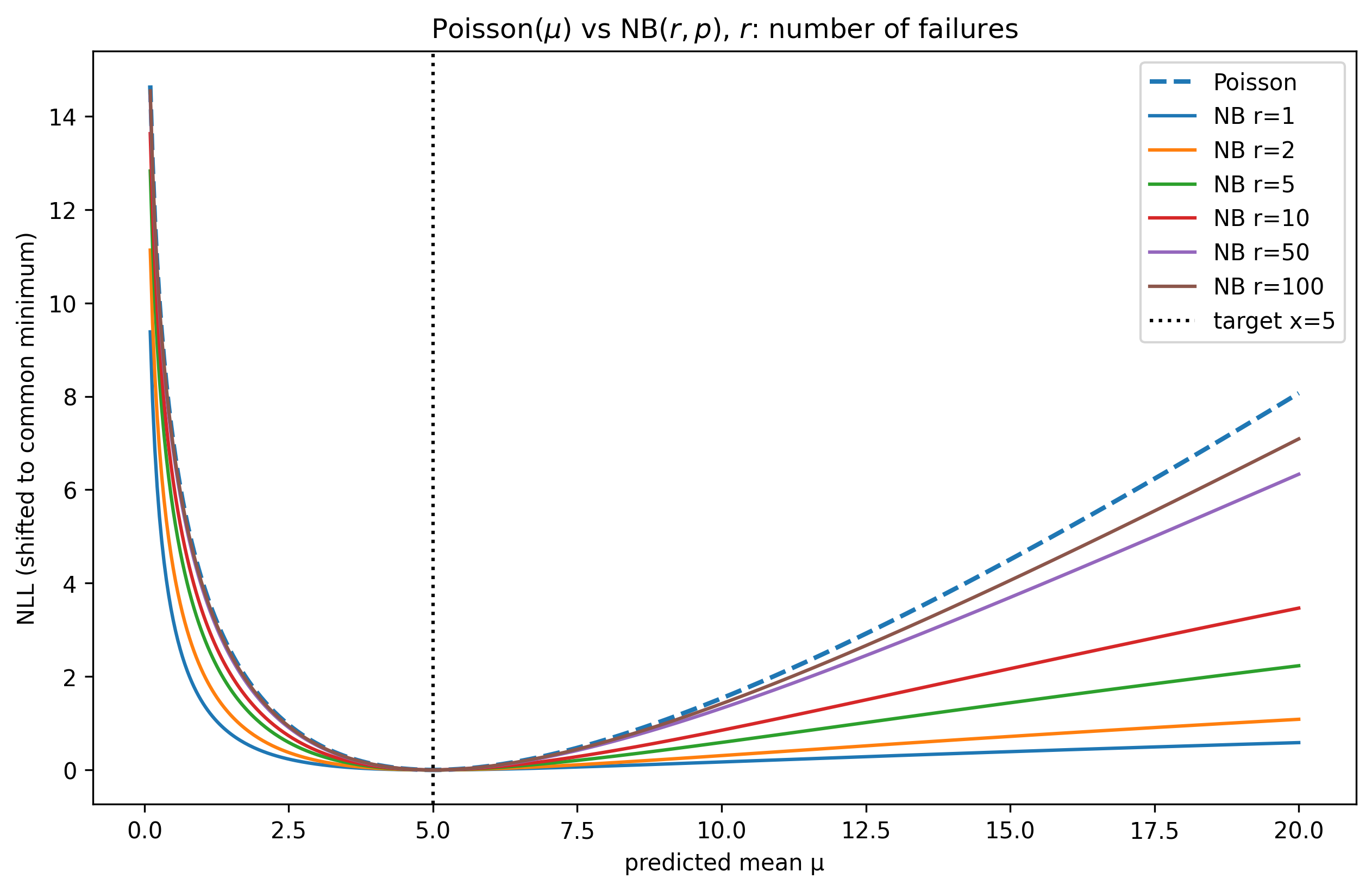
Fig 7: Poisson vs NB NLL for a single observation and a range of r values for NB. The mean
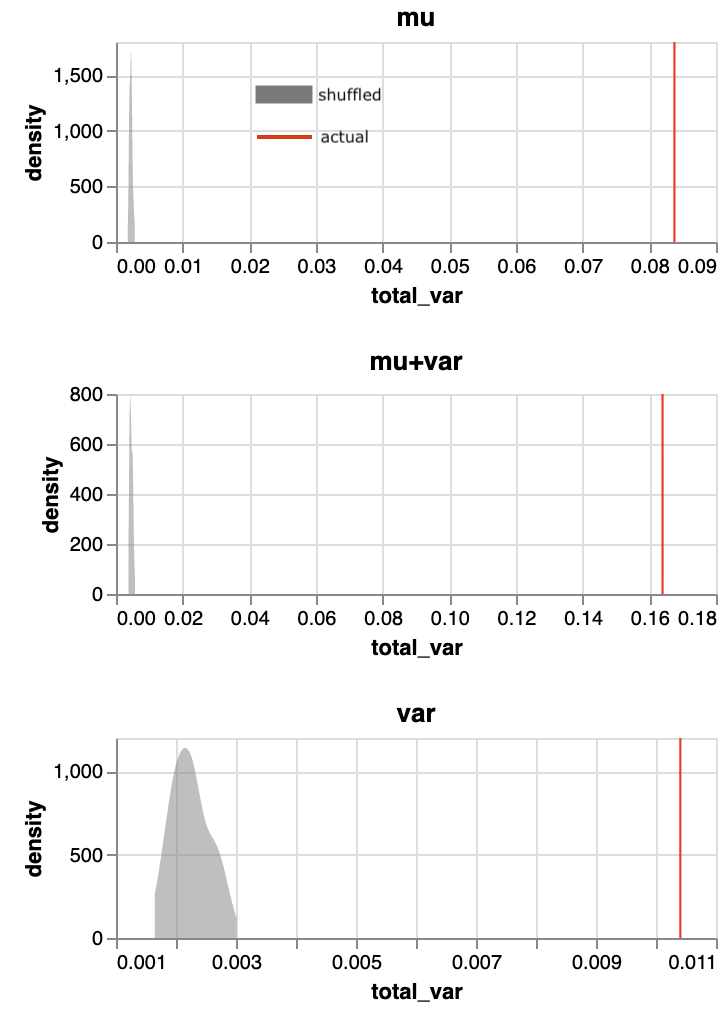
Fig 8: ZINB vs NB NLL for a single observation and a range of r values for NB and π values
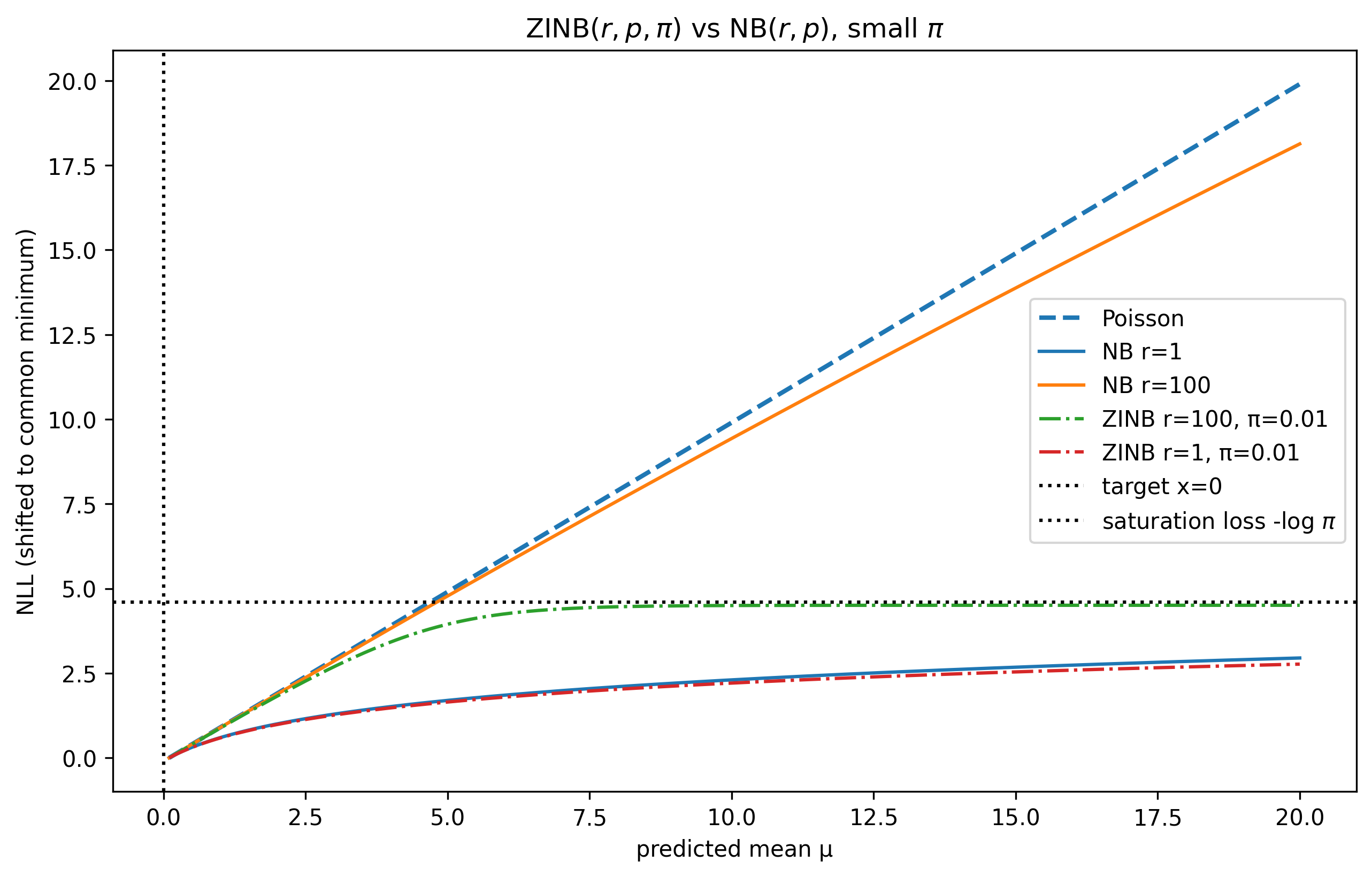
Fig 9: Variance explained captured by gene mean and gene variance against principal com-
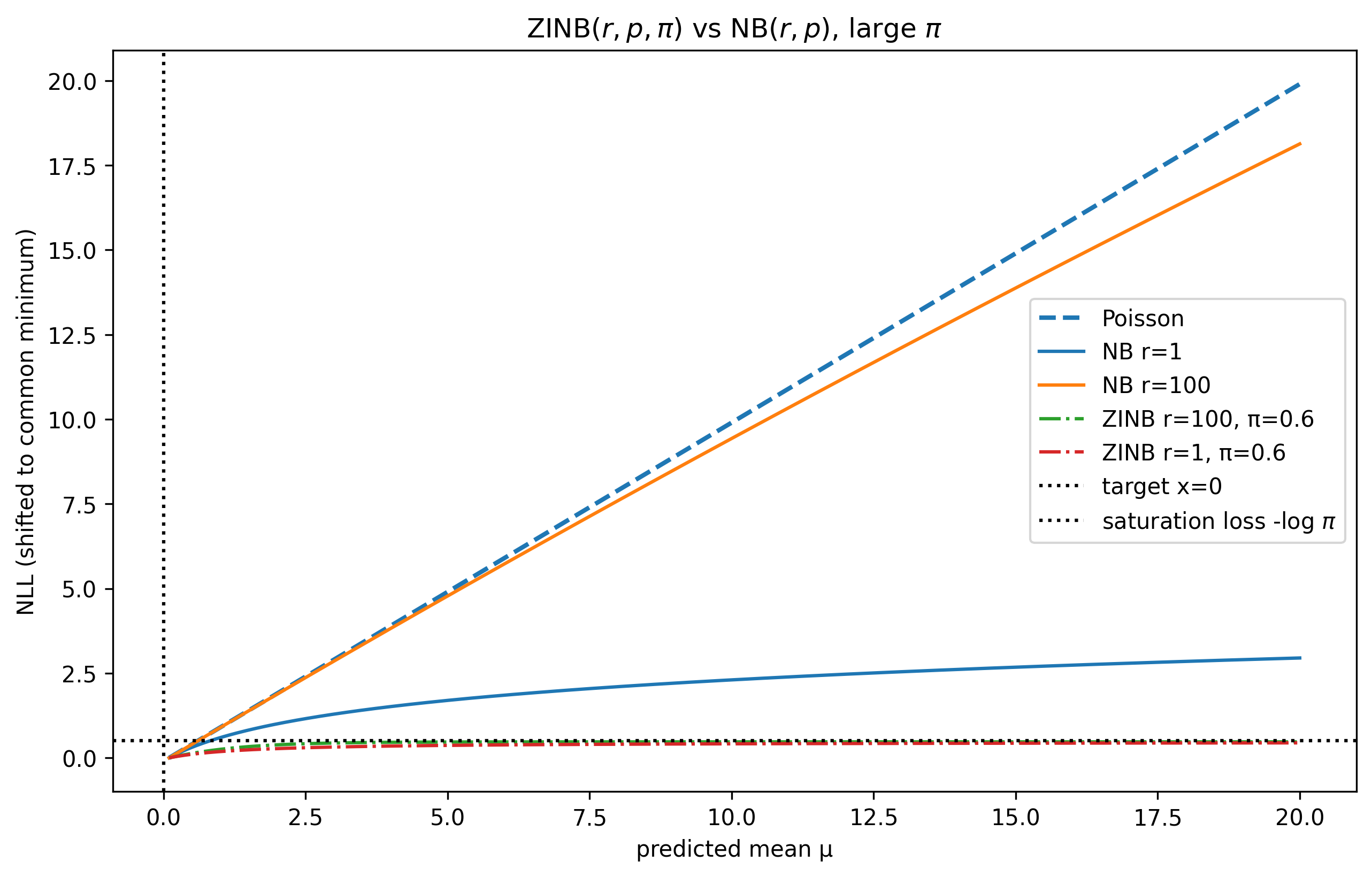
Fig 4 (page 21).
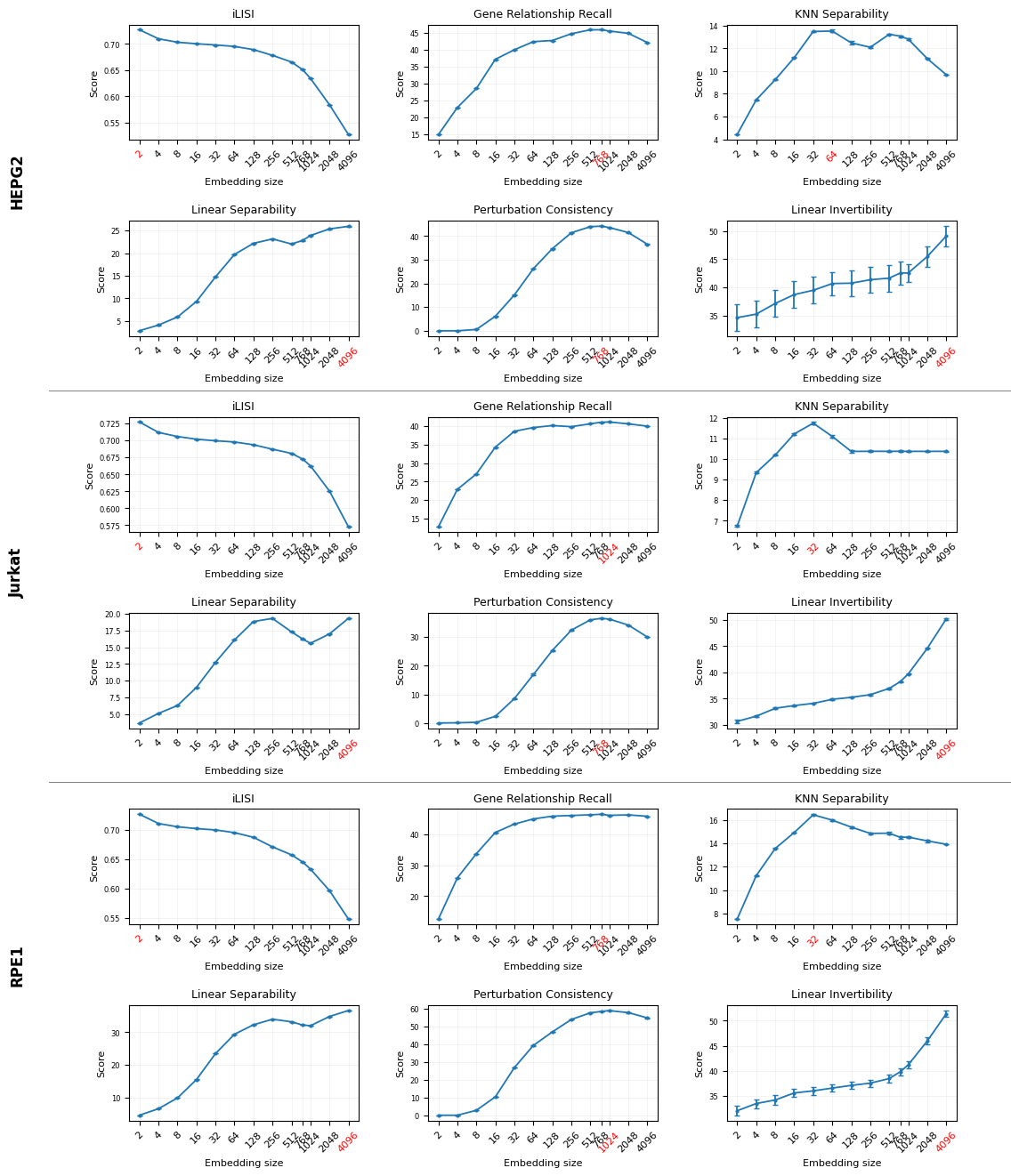
Fig 11: Dimension size ablation for PCA. Bendidi et al. (2024) benchmarking results on RPE1,
Limitations
- Model trained and evaluated primarily on oncology/perturbation-focused datasets; generalization to other tissues or conditions is untested and distributionally biased.
- No evaluation against adversarial or malicious perturbations; robustness to adversarial attacks remains unknown.
- Scaling laws for transcriptomic SSL models for larger data and architectures are not fully explored; large gains beyond proposed architecture are uncertain.
- The assembled DiverseRNA-1.4M dataset, while public, involves substantial curation steps that may not be trivially reproducible without authors’ pipelines.
- Model focuses on gene expression counts only, excluding multi-omic or spatial modalities which may enrich biological signal.
- Evaluation predominantly uses predefined gene relationships and perturbation benchmarks; discovery of novel biological mechanisms remains to be demonstrated.
Open questions / follow-ons
- Can the TxFM masked autoencoding approach be extended to integrate multi-modal data such as protein, chromatin, or spatial transcriptomics to further improve representation learning?
- How do scaling laws for model size, dataset size, and architecture complexity affect performance and transfer learning in transcriptomics compared to findings here?
- Can the learned gene embeddings from TxFM be mined to discover previously unknown gene functional relationships or drug targets in prospective biological validation?
- How robust is TxFM to batch effects and technical noise across very diverse RNA-seq modalities and platforms beyond the oncology-focused training datasets?
Why it matters for bot defense
For bot-defense and CAPTCHA practitioners, this paper showcases a domain where large-scale self-supervised learning must carefully tailor architecture, loss, and data curation to extract meaningful representations from noisy high-dimensional count data. Similar principles may apply to CAPTCHA signals where raw features are noisy and high-dimensional, implying that off-the-shelf large foundation models without inductive biases or domain-specific design may underperform simple baselines. The emphasis on masking strategies, loss functions respecting data distributions, and curation to improve signal over scale offers lessons for designing effective CAPTCHAs or bot defense embeddings that generalize robustly. Moreover, the finding that learned representations encode rich relationships on low-dimensional manifolds suggests that linear or PCA postprocessing might amplify signal in engineered CAPTCHA features. While transcriptomics is a different modality, the care taken in model-data synergy and transfer learning evaluation is directly applicable to security systems where robustness to unseen inputs and interpretability of learned features matter.
Cite
@article{arxiv2605_31562,
title={ Effective Biological Representation Learning by Masking Gene Expression },
author={ Kian Kenyon-Dean and Alina Selega and Ihab Bendidi and Jordan M. Sorokin and Luca Bertinetto and David Errington and Hayley Donnella and Oren Kraus },
journal={arXiv preprint arXiv:2605.31562},
year={ 2026 },
url={https://arxiv.org/abs/2605.31562}
}