
All-Electron Relativistic Fully Self-Consistent $GW$ Study of Heteronuclear Actinide-Containing Diatomics
Source: arXiv:2605.31571 · Published 2026-05-29 · By Jacob Adamski, Vibin Abraham, Dominika Zgid
TL;DR
This paper addresses the challenge of accurately modeling electronic structure and spectroscopic properties of uranium-containing diatomic molecules (UC, UN, UO, UF), which are archetypal actinide systems with strong relativistic and electron correlation effects. Traditional high-level wavefunction methods with explicit treatment of spin–orbit coupling (SOC) are accurate but computationally expensive. The authors present the first fully self-consistent all-electron GW (scGW) calculations combined with the exact two-component (X2C) relativistic Hamiltonian framework, which includes scalar relativistic and variational SOC effects. They provide systematic benchmarks of adiabatic ionization potentials (AIP), adiabatic electron affinities (AEA), vertical detachment energies (VDE), equilibrium bond lengths, and harmonic vibrational frequencies for these actinide diatomics, comparing to experiment and state-of-the-art wavefunction methods such as Feller–Peterson–Dixon (FPD) composite and CASPT2 approaches.
The results demonstrate that scGW with X2C provides a practical, starting-point–independent, and variationally relativistic many-body Green's function approach delivering ionization energies and vibrational properties in very good agreement with high-level benchmarks and experiments, with errors generally below 0.1–0.2 eV for ionization potentials and vibrational frequencies within 10–20 cm–1. For electron affinities and detachment energies, diffuse basis sets are crucial due to the spatially extended nature of the anionic states. UF is identified as a particularly challenging test case needing full two-component spinor treatment of SOC; scalar relativistic methods significantly underpredict electron attachment/detachment energies, while X2C-scGW recovers agreement with experiment and high-level CCSD(T) results. This work establishes all-electron X2C-scGW as an accurate and scalable route for computational actinide spectroscopy and motivates its application to larger uranium-containing molecules and materials.
Key findings
- All-electron X2C-scGW predicts adiabatic ionization potentials (AIPs) of UC, UN, UO, and UF within 0.03–0.16 eV of experimental or FPD benchmark values after CBS extrapolation (Table II).
- Scalar relativistic scGW (sfX2C) overestimates AIPs by up to ~0.2 eV for UC, UN, UO; including spin–orbit coupling through full two-component X2C reduces these errors by roughly half.
- Diffuse augmented basis sets increase the computed adiabatic electron affinities (AEA) by ~0.03–0.08 eV in scGW calculations, necessary to converge weakly bound anionic states (Table III).
- scGW systematically underestimates AEAs and VDEs by ~0.1–0.2 eV relative to FPD and experiment, primarily due to missing higher-order correlation beyond GW.
- For UF, scalar relativistic scGW underestimates VDE and AEA by ~0.2–0.25 eV, while X2C-scGW incorporating variational spin–orbit coupling improves agreement to within 0.14 eV of experiment (Fig. 2).
- Natural orbital analysis shows uranium valence configurations of 5f27s1 for UC/UN, 5f37s1 for UO, and 5f37s2 for UF, consistent with prior theoretical studies (Table I).
- scGW predicts equilibrium bond lengths and harmonic vibrational frequencies close to experimentally measured and CCSD(T) values, with underestimation of bond lengths by ~0.01–0.03 Å in most cases (Table V).
- Self-consistency convergence for scGW with DIIS is achievable for these challenging open-shell actinide molecules, with chemical potential reoptimized each iteration (Fig. 1).
Methodology — deep read
Threat model & assumptions: This is a computational chemistry study rather than a security paper, so no attacker model is defined. The goal is to test accuracy and convergence of fully relativistic self-consistent GW methods on actinide-containing molecules, comparing to experiments and high-level quantum chemistry. Assumed exact nuclear geometries or accurate correlated geometries from literature were used.
Data: The molecules studied are small uranium diatomics (UC, UN, UO, UF). Experimental geometries were used when available; otherwise geometries obtained from SO-CASPT2 calculations. The electronic structure calculations employed correlation-consistent basis sets designed for relativistic Hamiltonians: cc-pVnZ-DKH3 for uranium, cc-pVnZ or aug-cc-pVnZ for ligands (C, N, O, F). Triple- and quadruple-zeta quality basis sets (TZ, QZ) were used, with complete basis set (CBS) extrapolation for improved accuracy. Diffuse augmented basis sets were included for electron affinity and detachment calculations to capture diffuse anionic orbitals.
Architecture / algorithm: The method uses fully self-consistent GW (scGW), a Green’s function many-body perturbation theory approach iteratively solving Dyson’s equation for the one-particle Green’s function and self-energy. Relativistic effects are included variationally via the exact two-component (X2C) Hamiltonian, allowing both scalar relativistic corrections and spin–orbit coupling (SOC) on the same footing. The dynamic self-energy is computed on the imaginary Matsubara frequency/time axis using resolution-of-identity and intermediate representation for computational efficiency. The computed Green’s function and self-energy matrices are represented in atomic orbital basis.
Training regime: Not applicable. Energy and property calculations were performed using the 'green-mbpt' code. DIIS convergence acceleration was applied with a modified error vector based on Fock matrix and density matrix commutators. The chemical potential was updated at each iteration to maintain correct particle number. Calculations were done at inverse temperature β=1000 a.u.–1. CBS extrapolation was performed via a two-point formula using TZ and QZ results.
Evaluation protocol: Quantities computed include adiabatic ionization potentials (AIP), electron affinities (AEA), vertical detachment energies (VDE), equilibrium bond lengths (re), and harmonic vibrational frequencies (ωe). Results were compared against experimental data and benchmark theoretical methods including FPD composite coupled cluster and SO-CASPT2. Basis-set convergence and relativistic treatment effects were carefully analyzed by comparing scalar relativistic (sfX2C) and fully relativistic (two-component X2C) results. The impact of diffuse basis functions on anionic properties was also tested. Convergence of scGW iterations was monitored.
Reproducibility: The authors used versions of PySCF (2.2.1) for mean-field and integrals and their in-house 'green-mbpt' code for GW calculations. Details on basis sets and geometries are provided, with some supporting info for potential energy curves and two-electron relativistic corrections. Though no direct code release or frozen weights are stated, the use of standard basis sets and software suggest reproducibility is possible within computational chemistry norms. Some data (e.g. experimental geometries) come from public literature.
Concrete example end-to-end: For the UN molecule, the scGW iteration was started with a PBE0 hybrid functional reference for sfX2C and a generalized Hartree-Fock (GHF) reference for full X2C. A damping factor was applied in the first five iterations for stability. The chemical potential was updated each iteration to enforce correct electron count. The self-energy was computed in imaginary time/frequency space and the Dyson equation solved iteratively until energy and norm convergence below 1e–5 a.u. was reached (Fig. 1). Resulting natural orbitals and AO populations were extracted for analysis (Table I). Ionization potentials and vibrational properties were then computed at the relaxed geometries and compared to experiment and other calculations.
Technical innovations
- First application of an all-electron fully self-consistent GW (scGW) method combined with exact two-component (X2C) relativistic Hamiltonian to open-shell uranium-containing molecules.
- Use of variational two-component relativistic treatment including spin–orbit coupling within the scGW framework, improving accuracy for actinide electron attachment/detachment processes.
- Integration of sparse imaginary-time/frequency intermediate representation grids and modified DIIS acceleration to achieve practical convergence for challenging actinide scGW calculations.
- Systematic assessment of basis set effects, establishing critical role of diffuse basis functions on ligand atoms for converging electron affinities and vertical detachment energies in actinide diatomics.
Datasets
- UC, UN, UO, UF uranium diatomic molecules — ~4 small heteronuclear diatomic systems — geometries from experiment or SO-CASPT2 calculations
Baselines vs proposed
- HF: AIP underestimates experiment by ~0.7 eV, scGW reduces error to 0.03–0.16 eV (Table II)
- SF-X2C-scGW: AIP error vs experiment up to 0.23 eV, two-component X2C-scGW reduces error to as low as 0.03 eV
- scGW (sfX2C) AEA underestimates FPD benchmark by ~0.1–0.3 eV, addition of diffuse functions increases AEA by up to 0.08 eV (Table III)
- scGW (sfX2C) VDE underestimates experimental/FDP by ~0.18–0.24 eV; augmented basis sets improve values (Table IV)
- For UF, spin-free scGW VDE = 0.432 eV (underestimate by 0.2 eV), full X2C-scGW VDE = 0.765 eV (within 0.14 eV of experiment 0.63 eV) (Fig. 2)
- scGW vibrational frequencies within ~10–20 cm–1 of CCSD(T) and experimental values; bond lengths underestimated by 0.01–0.03 Å (Table V)
Figures from the paper
Figures are reproduced from the source paper for academic discussion. Original copyright: the paper authors. See arXiv:2605.31571.
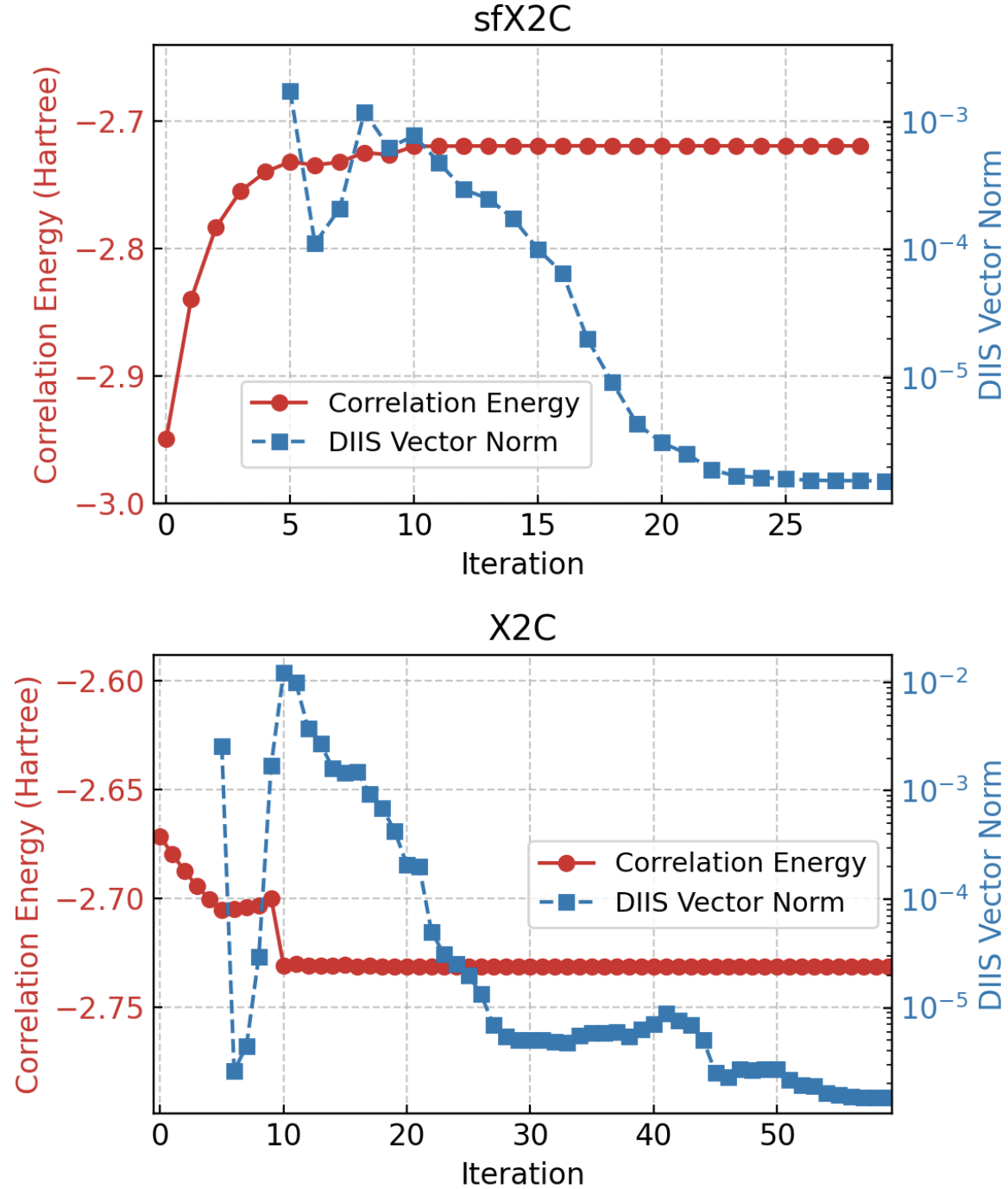
Fig 1: Convergence of scGW iterations for the UN system.
Limitations
- The GW approximation does not include higher-order correlation effects such as triples, resulting in systematic underestimation of electron affinities and vertical detachment energies compared to coupled cluster benchmarks.
- Basis set convergence remains challenging, especially for electron affinities requiring diffuse functions; CBS extrapolations help but residual incompleteness errors remain.
- Two-electron picture-change contributions in the X2C Hamiltonian are approximated, potentially leading to residual relativistic errors.
- Adiabatic nuclear gradients and full geometry optimizations within fully self-consistent GW are not yet implemented; geometries rely on external calculations or experiments.
- Spin–orbit coupling effects are treated variationally but only at two-component level; full four-component relativistic treatment could further improve accuracy but is currently too costly.
- UF represents a particularly difficult system with sensitivity to relativistic and correlation effects, indicating that limitations could be more pronounced for larger or more complicated actinides.
Open questions / follow-ons
- How well does fully self-consistent X2C-GW scale and perform on larger uranium- and actinide-containing polyatomic molecules or clusters beyond diatomics?
- Can higher-order vertex corrections beyond GW, or embedding approaches, be combined with X2C-scGW to systematically improve treatment of correlation effects and reduce systematic underestimation of electron affinities?
- What is the impact of two-electron relativistic corrections and possible four-component relativistic extensions on accuracy for heavy-element systems within GW frameworks?
- How can adiabatic nuclear gradients and full geometry optimization be incorporated practically within fully self-consistent two-component GW to enable predictive vibrational spectroscopy for actinide complexes?
Why it matters for bot defense
While this paper does not directly address bot defense or CAPTCHA technology, the underlying methodological advances in accurate, scalable, relativistic electronic structure prediction illustrate the potential of modern many-body Green's function techniques combined with state-of-the-art numerical methods. Bot defense researchers aiming to deploy machine learning or spectral analysis models in complex environments could draw inspiration from the authors' treatment of complex, strongly correlated, and highly relativistic systems, especially regarding convergence acceleration, basis set completeness, and variational treatment of complex effects (e.g., spin–orbit coupling). Moreover, the framework demonstrated for systematically improving predictions through variational and basis set enhancements may provide conceptual parallels for developing robust and accurate bot-detection algorithms under adversarial or noisy conditions. However, direct application to CAPTCHA or bot detection would require very domain-specific adaptation.
Cite
@article{arxiv2605_31571,
title={ All-Electron Relativistic Fully Self-Consistent $GW$ Study of Heteronuclear Actinide-Containing Diatomics },
author={ Jacob Adamski and Vibin Abraham and Dominika Zgid },
journal={arXiv preprint arXiv:2605.31571},
year={ 2026 },
url={https://arxiv.org/abs/2605.31571}
}