
ENSEMBITS: an alphabet of protein conformational ensembles
Source: arXiv:2605.13789 · Published 2026-05-13 · By Kaiwen Shi, Carlos Oliver
TL;DR
This paper addresses the limitation of existing protein structure tokenizers (PSTs) that only capture static local geometry but fail to encode protein dynamics reflected by conformational ensembles. The authors introduce ENSEMBITS, the first protein tokenizer designed specifically for conformational ensembles, which maps variable-sized unordered sets of structural frames into discrete tokens capturing correlated residue motions. ENSEMBITS solves challenges of permutation invariance, variable ensemble sizes, and sparsity of dynamic data by using SE(3)-invariant per-residue descriptors aggregated via a permutation-invariant set encoder and quantized with a Residual Vector-Quantized VAE. A novel single-frame-to-token distillation enables dynamics token prediction from a single static structure, addressing data sparsity. Trained on a large molecular dynamics corpus (mdCATH-div and MISATO), ENSEMBITS outperforms baselines in RMSF prediction, encodes meaningful dynamic motifs, and matches or exceeds static tokenizers on functional tasks such as enzyme classification, GO term, binding site/affinity prediction, and zero-shot mutation effect prediction, despite using less pretraining data. This discrete dynamics-aware alphabet unlocks new potential for protein language modeling and design by capturing functional relevant ensemble dynamics.
Key findings
- ENSEMBITS achieves 0.604 ± 0.001 Spearman correlation in RMSF prediction on mdCATH-div (P=full), beating the best baseline ProtProfileMD_K (0.579) by ∼2.5 points.
- For single-frame inputs (P=1), ENSEMBITS attains 0.458 Spearman on mdCATH-div RMSF, surpassing the second-best single-frame baseline AminoAseed (0.408) by about 5 points.
- Single-frame-to-token distillation reduces dynamics data requirement by enabling dynamics token prediction from one predicted static structure, validated by the P=1 model performance.
- ENSEMBITS tokens correlate strongly with residue motion amplitude, dominating token-conditioned ANOVA tests over other methods (quantitative details in Appendix E).
- On the zero-shot protein mutation effect prediction benchmark (PROTEINGYM, 96 DMS assays), combining ENSEMBITS tokens with ESM2 embeddings improves mean Spearman correlation by +6.9% over ESM2 alone, outperforming other structural tokenizers blended similarly.
- ENSEMBITS matches or exceeds ESM3struct and other baselines on multiple MISATO functional tasks (EC classification, GO term prediction, binding site and affinity prediction), particularly excelling in binding-site AP with skill scores up to 2.08 and AUROC up to 1.41 on structure splits.
- Partial ensembles (P=1) achieve comparable functional task performance to full ensembles (P=full), demonstrating the effectiveness of the distillation objective.
- The learned 2048-entry codebook encodes interpretable local motifs spanning rigid (low-motion) and flexible (high-motion) residue environments, validated by exemplar superposition visualizations.
Threat model
n/a — The paper focuses on protein dynamics representation for machine learning and functional prediction, rather than adversarial or security threats.
Methodology — deep read
The authors define the protein dynamics tokenization problem as mapping an unordered multiset of P protein conformations' per-residue geometric descriptors to discrete tokens representing correlated local motions.
Threat Model & Assumptions: The adversary model is not security-focused but the design assumes dynamics data from molecular dynamics (MD) simulations or predicted ensembles and tackles sparsity inherent in dynamics data. It does not assume access to ensembles at inference, supporting single-frame queries.
Data: Training data combines mdCATH-div (∼5,400 CATH domains with multiple temperatures and replicas) and MISATO (∼17,000 protein–ligand MD trajectories) with homology deduplication at the H-superfamily level to mitigate leakage. Each protein ensemble contains between 8–10 frames typically. Labels for evaluation include RMSF per residue, mutation effect data from PROTEINGYM, and multiple downstream function prediction benchmarks (EC, GO, binding sites).
Architecture / Algorithm: The core module is a Residual Vector-Quantized VAE (RVQ-VAE). Inputs are per-residue, per-frame SE(3)-invariant descriptors capturing neighbors' relative frames, encoding contact dynamics. The set encoder aggregates the P-frame multiset of descriptors into a single latent vector z via a PerceiverIO-inspired permutation-invariant cross-attention mechanism with learnable query tokens. This latent is quantized through a multi-level residual vector quantizer producing tuples of codebook indices (tokens). The decoder reconstructs the input multiset of descriptors from the quantized latent, trained with a Hungarian-matching permutation-invariant reconstruction loss.
Training regime: Models are trained end-to-end with losses including reconstruction, VQ commitment, and a novel single-frame-to-token distillation (SFTD) loss that aligns embeddings of subsampled single-frame inputs to full ensemble embeddings, allowing single-frame token prediction. Training hyperparameters are detailed in Appendices but involve standard deep learning practices (e.g., EMA codebook updates, cosine learning rate decay).
Evaluation protocol: Performance is assessed on RMSF prediction (Spearman correlation on held-out residues and sequences), token-dynamics correlation through ANOVA, zero-shot mutation effect scoring (Spearman on 96 assays), and functional classification/regression tasks on independent splits (sequence, structure, random clusters). Comparisons include multiple static and dynamic structural tokenizers. Multiple random seeds (10) and cross-validation strategies are used.
Reproducibility: Code is released openly. The training datasets are partially public (mdCATH-div, MISATO), though some corpus preprocessing details are deferred. Frozen weights availability is not explicitly stated. The authors provide detailed appendices describing model components, training, and evaluation.
Example end-to-end: For a protein ensemble with P=10 MD frames, per-residue descriptors are computed as relative SE(3) frames of neighbors per frame, aggregated by the permutation-invariant set encoder to a latent representation. This embedding is quantized via RVQ-VAE to a discrete token tuple representing the local dynamic motif. The decoder attempts to reconstruct the original multiset of descriptors, training with Hungarian-matched MSE loss. Single-frame-to-token distillation trains the model such that embeddings from subsampled single frames approximate full-ensemble embeddings, enabling token inference from one static predicted structure, subsequently used for downstream function prediction or mutation effect scoring.
Technical innovations
- Formulation of protein dynamics tokenization as a permutation-invariant compression of multiset per-residue SE(3)-invariant descriptors into discrete tokens.
- Use of a Residual Vector-Quantized Variational Autoencoder (RVQ-VAE) with multi-level codebooks to capture dominant dynamic modes while absorbing fine-grained variation.
- Design of a permutation-invariant set encoder inspired by PerceiverIO to aggregate unordered variable-size ensembles into single latent vectors.
- Introduction of single-frame-to-token distillation (SFTD) aligning single-frame embeddings with full-ensemble counterparts, enabling dynamics-aware token prediction from static structures.
Datasets
- mdCATH-div — ∼5,400 protein domains, 5 temperatures × 5 replicas — from mdCATH dataset, homology deduplicated at H-superfamily level
- MISATO — ∼17,000 protein–ligand complexes with MD trajectories — derived from PDBbind
Baselines vs proposed
- ProtProfileMD_K: RMSF Spearman = 0.579 on mdCATH-div vs ENSEMBITS P=full: 0.604
- AminoAseed: RMSF Spearman = 0.408 on mdCATH-div (single-frame) vs ENSEMBITS P=1: 0.458
- ESM3struct: mutation effect Spearman = 0.384 vs ENSEMBITS P=1: 0.381 (standalone), ENSEMBITS+ESM2 blend: 0.518 vs ESM2 alone: 0.484 (+6.9%)
- Binding-site prediction skill scores up to 2.08 (AP) and 1.41 (AUROC) on MISATO structure splits for ENSEMBITS (exceeding ESM3struct and others).
Figures from the paper
Figures are reproduced from the source paper for academic discussion. Original copyright: the paper authors. See arXiv:2605.13789.
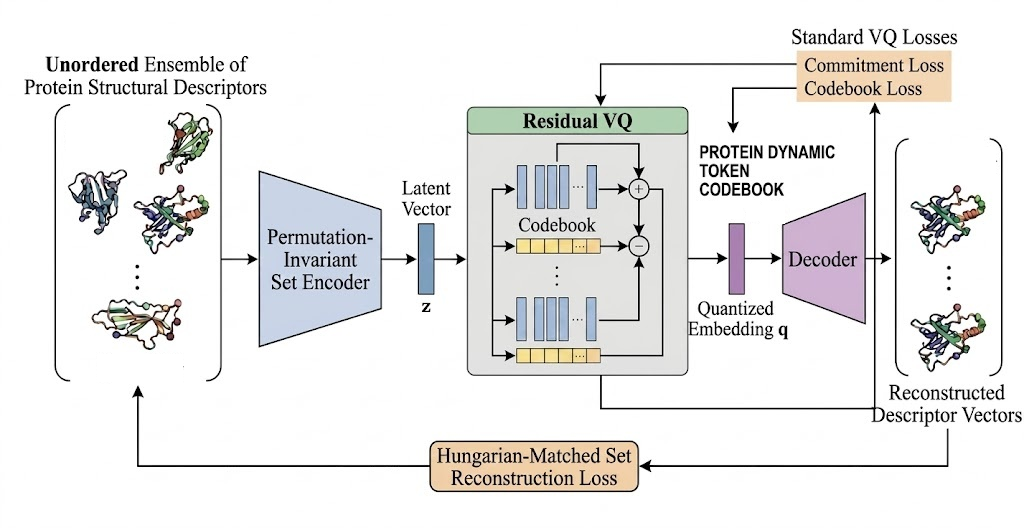
Fig 1: The Ensembits tokenization pipeline. The P-frame per-residue descriptor multiset is
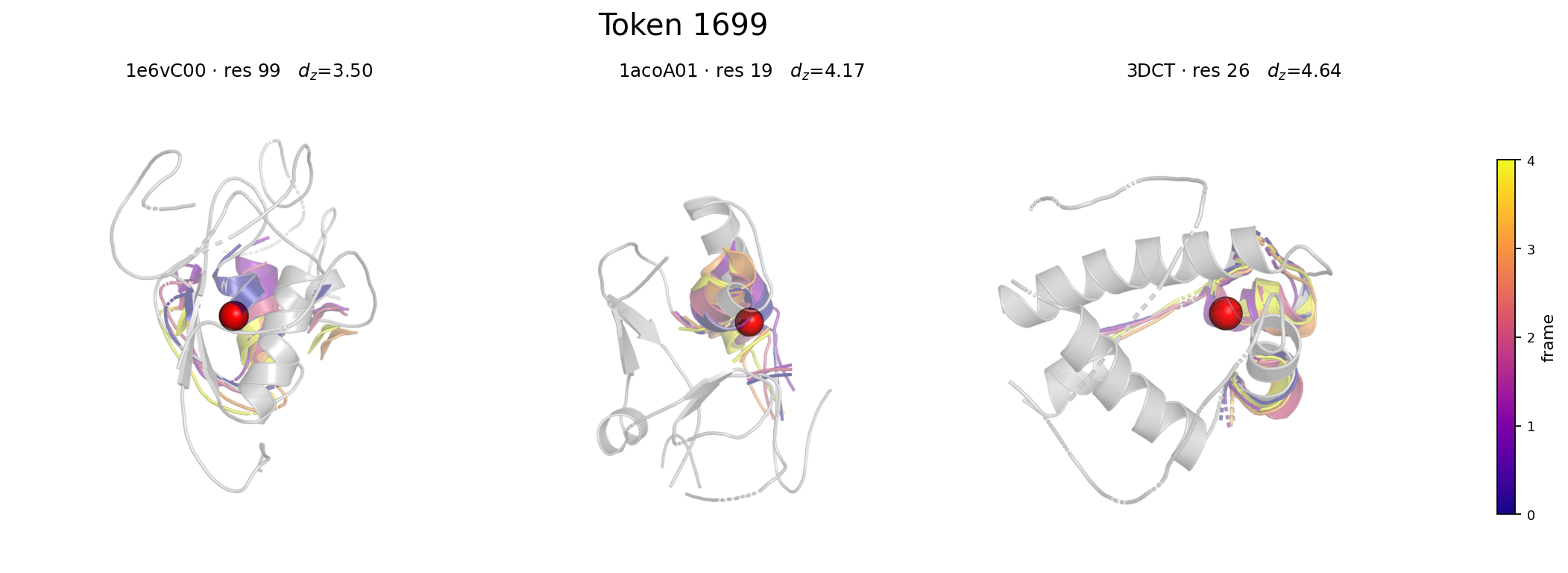
Fig 2: Token 1699 — a near-stationary local motif. Three distinct-protein exemplars (1e6vC00:99,
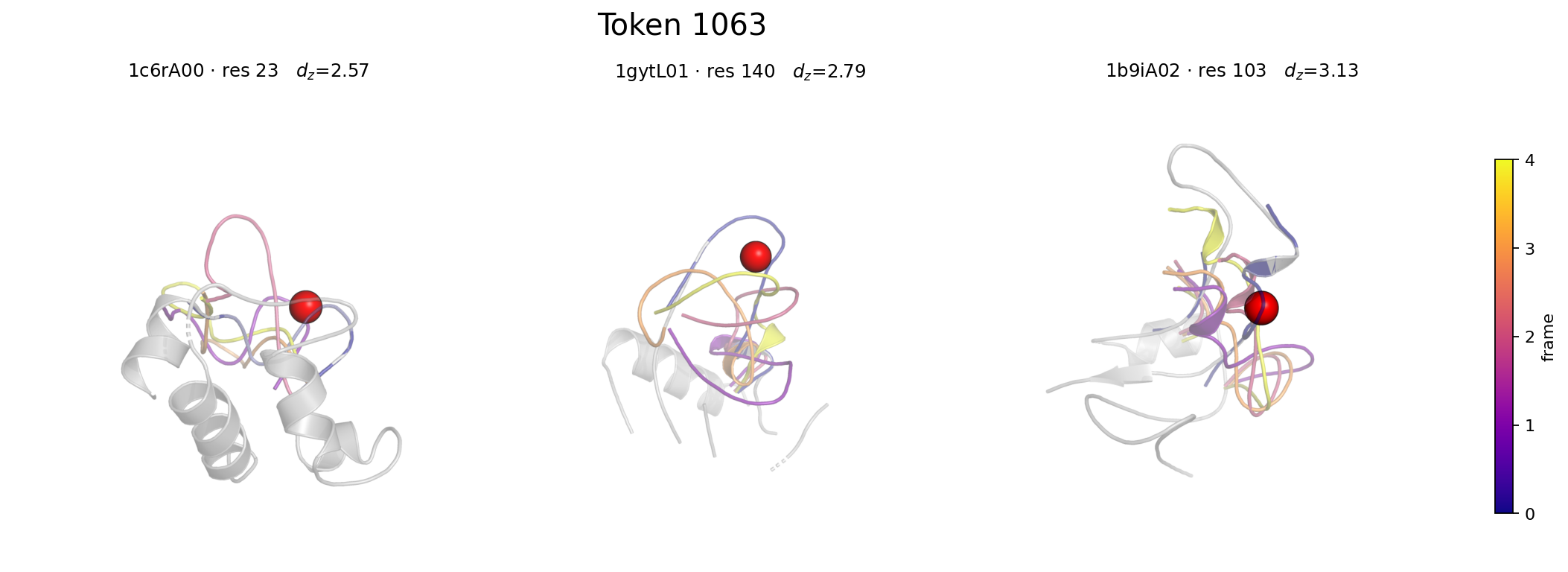
Fig 3: Token 1063 — a flexible local motif. Three exemplars (1c6rA00:23, 1gytL01:140,
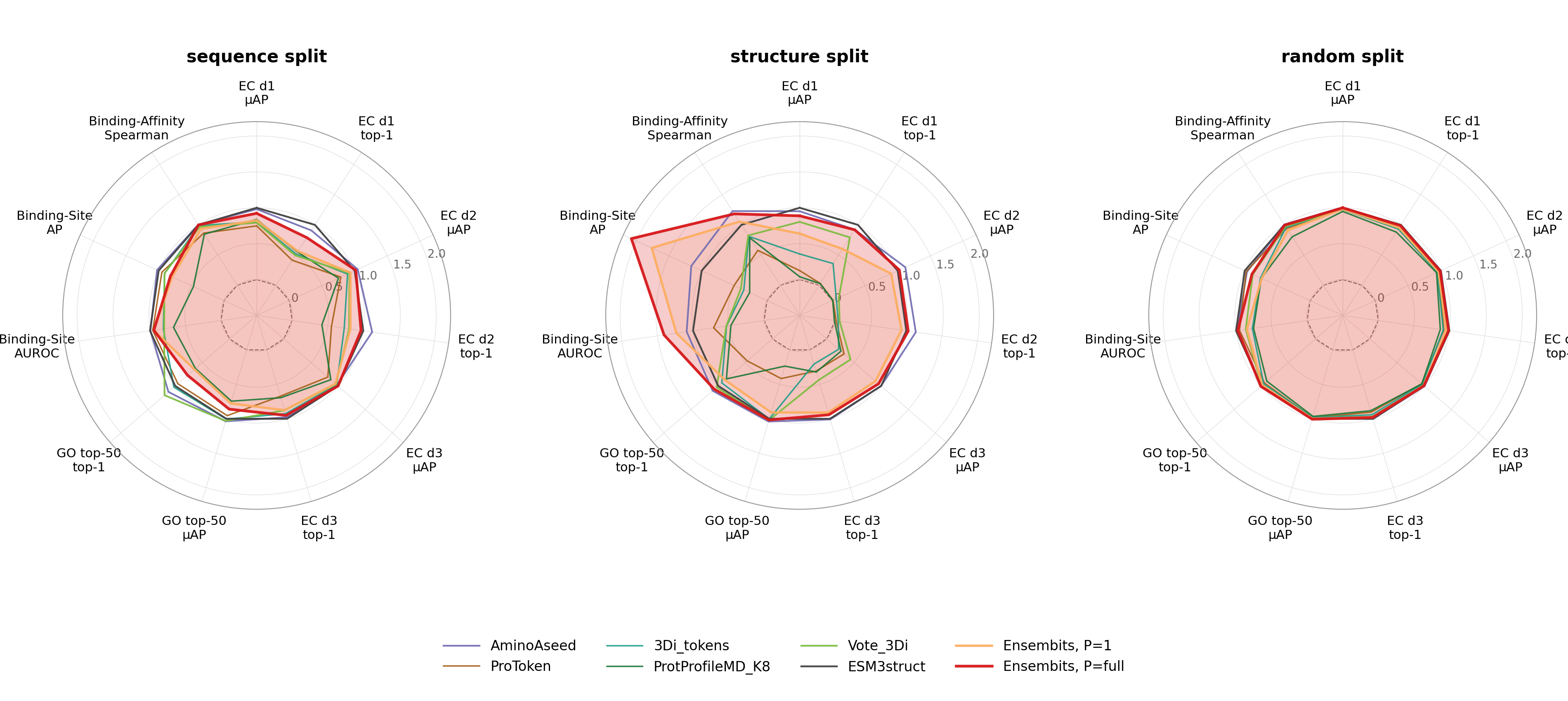
Fig 4: Skill-score radar across MISATO downstream tasks. Each panel is one ProteinShake
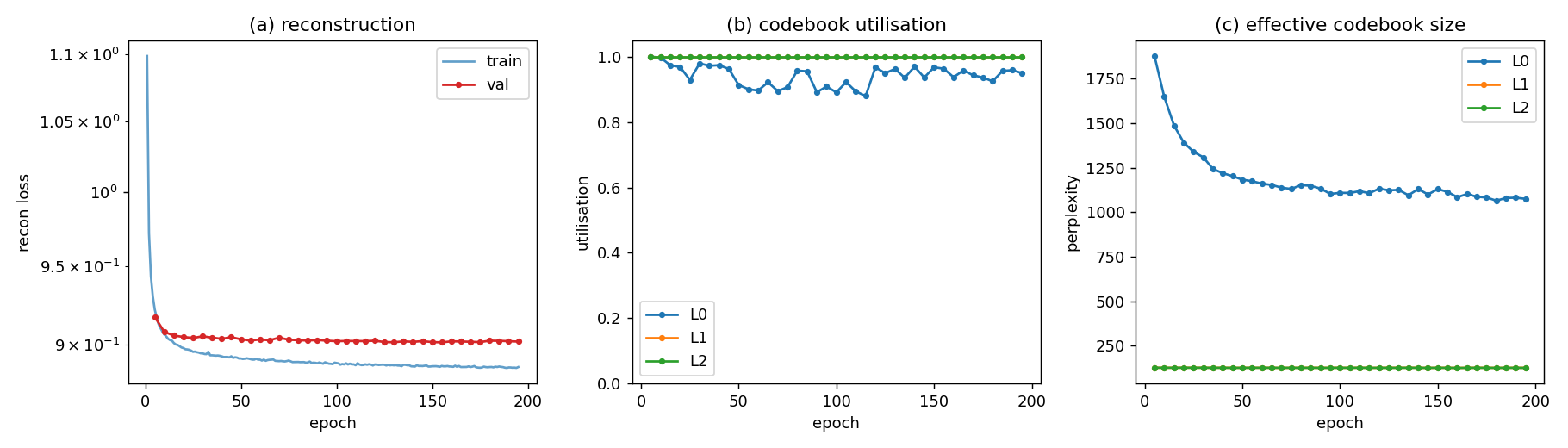
Fig 5: Training curve.
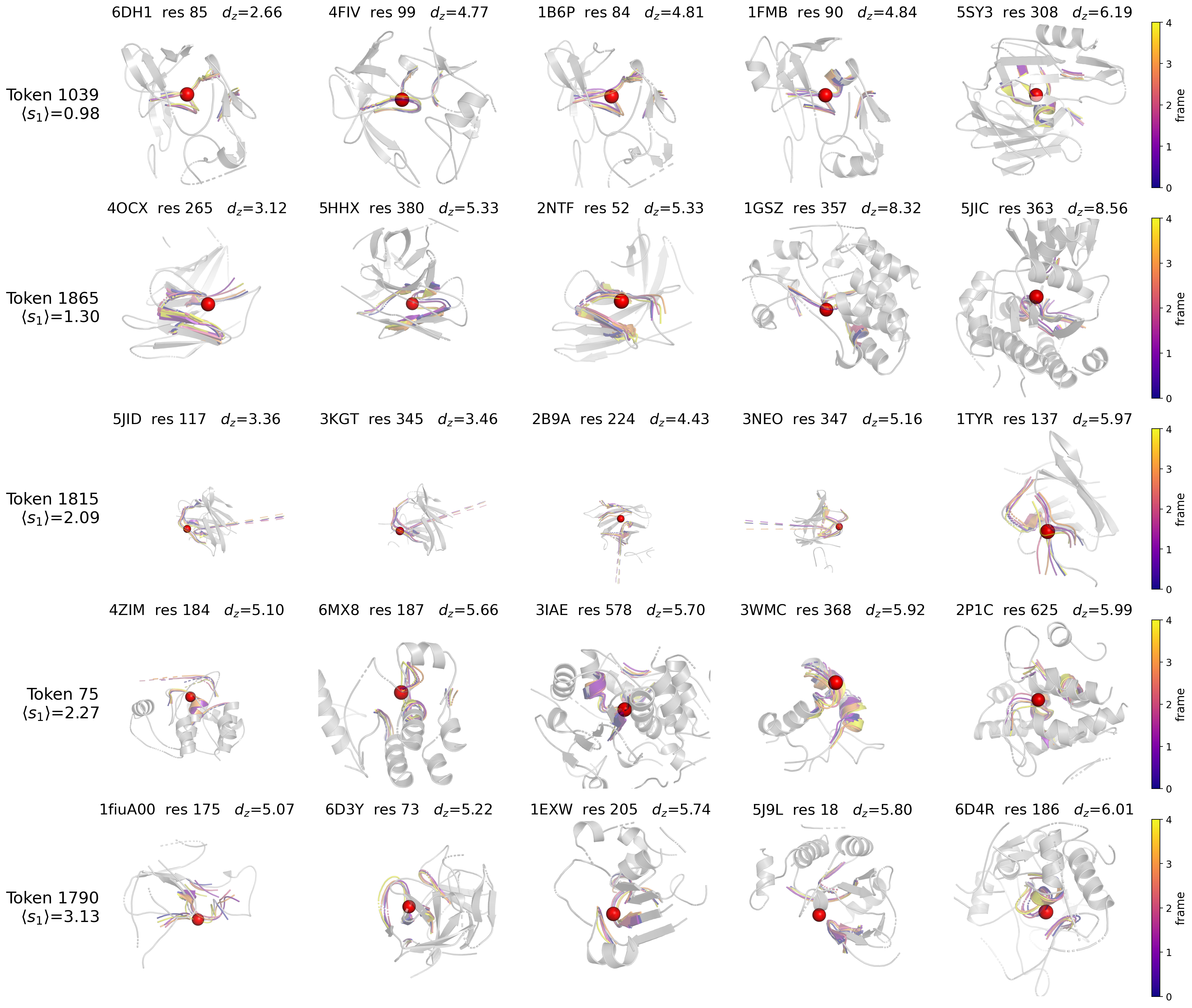
Fig 6: Five representative tokens from the codebook (rows), ordered top to bottom by increasing
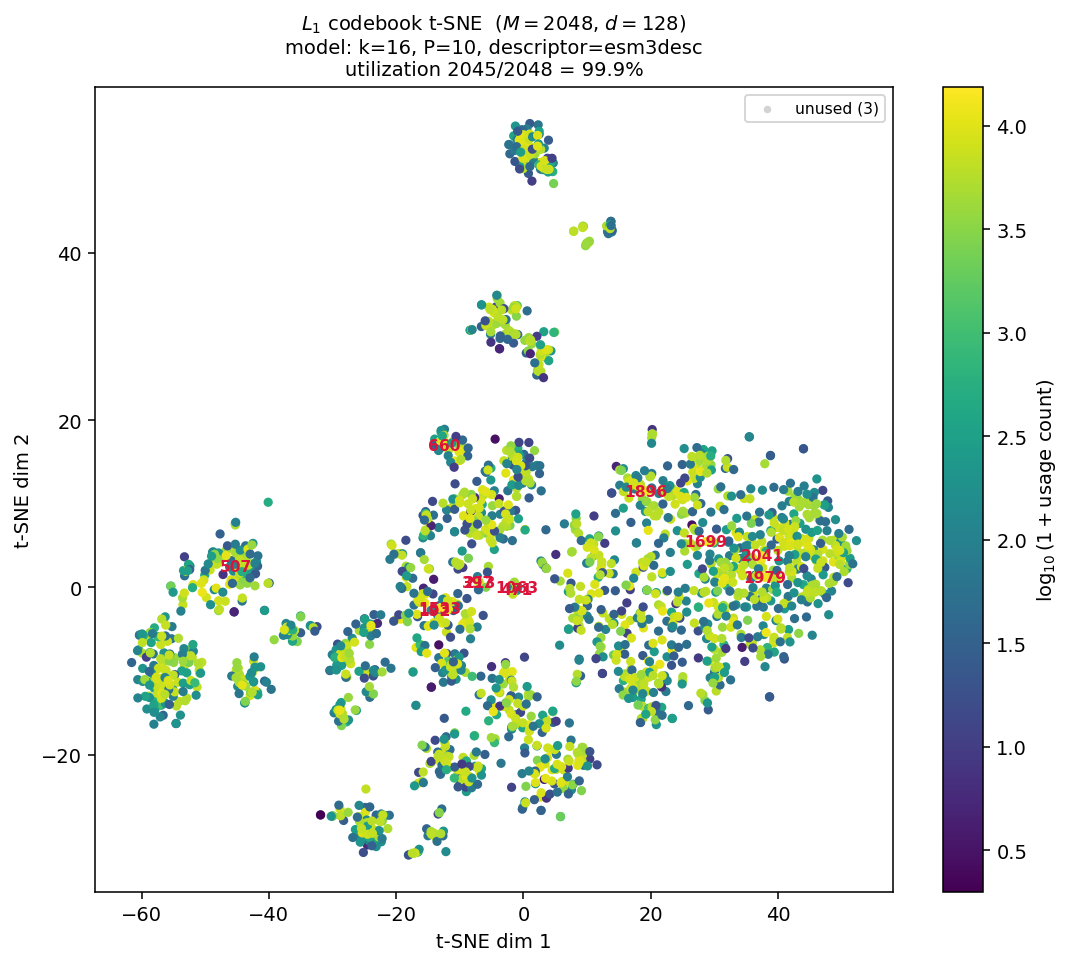
Fig 7: t-SNE projection of the L1 primary codebook (M = 2048 entries, d = 128) for the
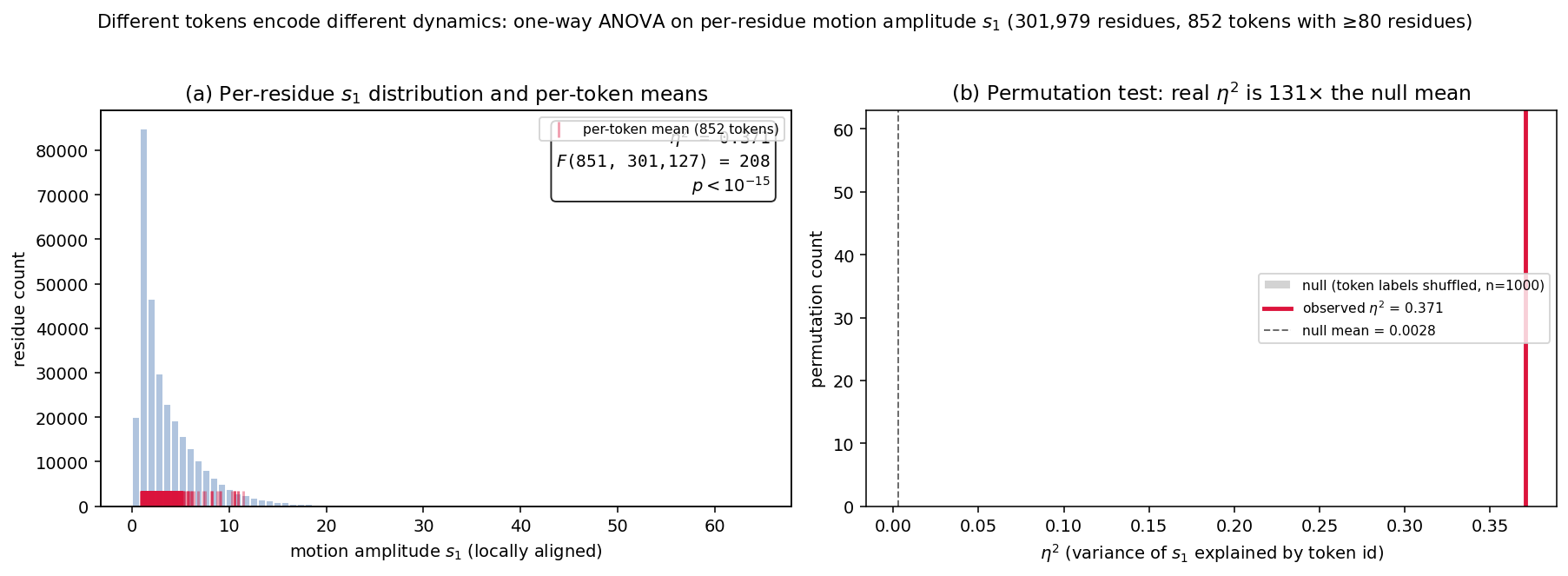
Fig 8: ANOVA test that different tokens encode different dynamics. Per-residue motion amplitude
Limitations
- Quality and coverage of learned tokens depend on the underlying MD trajectory datasets, which are costly and incomplete representations of real conformational space.
- Current ML-generated ensemble augmentation (e.g., AFSample2) is promising but limited by generator fidelity, affecting token quality.
- Functional validation focuses on prediction benchmarks but does not thoroughly test generalization to unseen folds or extreme motions.
- The model still requires single predicted static structures or ensembles at inference; completely sequence-only or low-data regimes remain challenging.
- The vocabulary size (2048 primary codewords) and residual quantization levels might limit granularity of dynamics captured, though this was not exhaustively explored.
- Decoder reconstruction depends on Hungarian matching, which may be computationally intensive for very large ensembles.
Open questions / follow-ons
- How does the ENSEMBITS vocabulary and learned token space evolve when scaling to larger, higher-quality ensemble datasets beyond current MD corpora?
- Can explicit dynamics tokens integrated into protein language models improve multi-frame structure generation or rational protein design?
- How well do ENSEMBITS tokens generalize to extreme conformational changes or rare states not represented in training data?
- What is the impact of choosing different geometric descriptor families or quantizer architectures on token informativeness and downstream performance?
Why it matters for bot defense
Though not a bot-defense or CAPTCHA-focused paper, ENSEMBITS demonstrates a novel method for compressing high-dimensional, unordered variable-size structural ensemble data into discrete tokens. This permutation-invariant, dynamics-aware tokenization approach could inspire strategies for summarizing complex, multi-modal input sequences or behaviors in other domains. The single-frame-to-token distillation method might suggest approaches for robust token prediction when full multi-frame data is unavailable, analogous to sparsity challenges in behavioral biometric analysis. Additionally, the use of Residual VQ-VAEs combined with permutation-invariant set encoders highlights promising architectures for representing complex structured inputs as discrete codes, potentially translatable to anomaly detection or bot behavior fingerprinting in traffic ensembles. Practitioners in bot-defense may find value in studying how rich time-series or ensemble data can be discretized without losing dynamic correlations.
Cite
@article{arxiv2605_13789,
title={ ENSEMBITS: an alphabet of protein conformational ensembles },
author={ Kaiwen Shi and Carlos Oliver },
journal={arXiv preprint arXiv:2605.13789},
year={ 2026 },
url={https://arxiv.org/abs/2605.13789}
}